10-Q: Quarterly report pursuant to Section 13 or 15(d)
Published on November 13, 2024
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-Q
(Mark One)
|
|
QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the quarterly period ended September 30, 2024
or
|
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
|
For the transition period from to
Commission file number: 001-35776
(Exact name of registrant as specified in its charter)
|
State of
|
|
|
|
(State or other jurisdiction of
incorporation or organization)
|
(I.R.S. Employer
Identification Number)
|
(Address of principal executive offices, including zip code)
(Registrant’s telephone number, including area code)
(Former name, former address, and former fiscal year, if changed since last report)
Securities registered pursuant to Section 12(b) of the Act:
|
Title of each class
|
Trading Symbol(s)
|
Name of each exchange on which registered
|
|
|
|
|
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for
such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter)
during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the
definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
|
Large accelerated filer
|
☐
|
Accelerated filer
|
☐
|
||
|
|
☒
|
Smaller reporting company
|
|
||
|
Emerging growth company
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting
standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The number of outstanding shares of common stock of the registrant, par value per share of $0.0001, as of November 13, 2024, was 10,139,861 .
GRACE THERAPEUTICS, INC.
(Formerly ACASTI PHARMA INC.)
QUARTERLY REPORT ON FORM 10-Q
For the Quarter Ended September 30, 2024
|
Page
|
||
|
PART I. FINANCIAL INFORMATION
|
||
|
Item 1.
|
5
|
|
|
Item 2.
|
20
|
|
|
Item 3.
|
37
|
|
|
Item 4.
|
37
|
|
|
PART II. OTHER INFORMATION
|
||
|
Item 1.
|
37
|
|
|
Item 1A.
|
38
|
|
|
Item 2.
|
38
|
|
|
Item 3.
|
38
|
|
|
Item 4.
|
38
|
|
|
Item 5.
|
38
|
|
|
Item 6.
|
38
|
|
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This quarterly report contains information that may be forward-looking statements within the meaning of U.S. federal securities laws and forward-looking statements within the meaning of
Canadian securities laws, both of which we refer to in this quarterly report as forward-looking statements. Forward- looking statements can be identified by the use of terms such as “may”, “will”, “should”, “expect”, “plan”, “anticipate”,
“believe”, “intend”, “estimate”, “predict”, “potential”, “continue” or other similar expressions concerning matters that are not statements about historical facts. Forward-looking statements in this quarterly report include, among other things,
information, or statements about:
|
|
• |
our ability to build a late-stage pharmaceutical company focused in rare and orphan diseases and, on developing and commercializing products that improve clinical outcomes using our novel drug delivery
technologies;
|
|
|
• |
our ability to apply new proprietary formulations to existing pharmaceutical compounds to achieve enhanced efficacy, faster onset of action, reduced side effects, and more convenient drug delivery that can
result in increased patient compliance;
|
|
|
• |
the potential for our drug candidates to receive orphan drug designation and exclusivity from the U.S. Food and Drug Administration
(“FDA”) or regulatory approval under the Section 505(b)(2) regulatory pathway under the Federal Food, Drug and Cosmetic Act (“FDCA”);
|
|
|
• |
the future prospects of our GTx-104 drug candidate, including but not limited to GTx-104’s potential to be administered to improve the
management of hypotension in patients with aneurysmal subarachnoid hemorrhage (“aSAH”); GTx-104’s potential to reduce the incidence of vasospasm in aSAH patients resulting in better outcomes; the ability of GTx-104 to achieve a
pharmacokinetic (“PK”) and safety profile similar to the oral form of nimodipine; GTx-104’s potential to provide improved bioavailability and the potential for reduced use of rescue therapies, such as vasopressors in patients with aSAH
the timing and outcome of the Phase 3 safety study for GTx-104; GTx-104’s potential to achieve pharmacoeconomic benefit over the oral form of nimodipine; GTx-104 formulation’s Chemistry Manufacturing and Control (CMC); GTx-104’s
Contract Manufacturing Organization (CDMO) to stay in compliance of FDA; our ability to ultimately file a new drug application (“NDA”) for GTx-104 under Section 505(b)(2) of the FDCA; the acceptance of the NDA by the FDA; and the timing
and ability to receive FDA approval for marketing GTx-104;
|
|
|
• |
our plan to prioritize the development of GTx-104;
|
|
|
• |
our plan to maximize the value of our de-prioritized drug candidates, GTx-102 and GTx-101, including through potential development, licensing, or sale of those drug candidates;
|
|
|
• |
the future prospects of our GTx-102 drug candidate, including but not limited to GTx-102’s potential to provide clinical benefits to decrease symptoms associated with Ataxia Telangiectasia; GTx-102’s
potential ease of drug administration; the timing and outcomes of a PK bridging study and Phase 3 efficacy and safety study for GTx-102; the timing of an NDA filing under Section 505(b)(2) in connection with GTx-102; and the ability to
receive FDA approval for marketing GTx-102;
|
|
|
• |
the future prospects of our GTx-101 drug candidate, including but not limited to GTx-101’s potential to be administered to postherpetic neuralgia (“PHN”) patients to treat the severe nerve pain associated
with the disease; assumptions about the biphasic delivery mechanism of GTx-101, including its potential for rapid onset and continuous pain relief for up to eight hours; and the timing and outcomes of single ascending dose/multiple
ascending dose and PK bridging studies, and a Phase 2 and Phase 3 efficacy and safety study; the timing of an NDA filing under Section 505 (b)(2) of the FDCA for GTx-101; and the timing and ability to receive FDA approval for marketing
GTx-101;
|
|
|
• |
the quality of our clinical data, the cost and size of our development programs, expectations and forecasts related to our target markets and the size of our target markets; the cost and size of our
commercial infrastructure and manufacturing needs in the United States, European Union, and the rest of the world; and our expected use of a range of third-party contract research organizations (“CROs”) and contract manufacturing
organizations (“CMOs”) at multiple locations;
|
|
|
• |
expectations and forecasts related to our intellectual property portfolio, including but not limited to the probability of receiving
orphan drug exclusivity from the FDA for our leading pipeline drug candidates; our patent portfolio strategy; and outcomes of our patent filings and extent of patent protection;
|
|
|
• |
our intellectual property position and duration of our patent rights;
|
|
|
• |
our strategy, future operations, prospects, and the plans of our management with a goal to enhance shareholder value our need for additional financing, and our estimates regarding our operating runway and
timing for future financing and capital requirements;
|
|
|
• |
our expectations regarding our financial performance, including our costs and expenses, liquidity, and capital resources;
|
|
|
• |
our projected capital requirements to fund our anticipated expenses; and
|
|
|
• |
our ability to commercialize GTx-104 in the United States or establish strategic partnerships or commercial collaborations or obtain non-dilutive funding.
|
Although the forward-looking statements in this quarterly report are based upon what we believe are reasonable assumptions, you should not place undue reliance on those forward-looking
statements since actual results may vary materially from them.
In addition, the forward-looking statements in this quarterly report are subject to a number of known and unknown risks, uncertainties and other factors, many of which are beyond our control,
that could cause our actual results and developments to differ materially from those that are disclosed in or implied by the forward-looking statements, including, among others:
|
|
• |
We are heavily dependent on the success of our lead drug candidate, GTx-104.
|
|
|
• |
Clinical development is a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results. Failure can occur at any stage of
clinical development.
|
|
|
• |
We are subject to uncertainty relating to healthcare reform measures and reimbursement policies that, if not favorable to our drug candidates, could hinder or prevent our drug candidates’ commercial
success.
|
|
|
• |
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell our drug products, if approved, we may be unable to generate any revenue.
|
|
|
• |
If we are unable to differentiate our drug products from branded reference drugs or existing generic therapies for similar treatments, or if the FDA or other applicable regulatory authorities approve
products that compete with any of our drug products, our ability to successfully commercialize our drug products would be adversely affected.
|
|
|
• |
Our success depends in part upon our ability to protect our intellectual property for our drug candidates.
|
|
|
• |
Intellectual property rights do not necessarily address all potential threats to our competitive advantage.
|
|
|
• |
We do not have internal manufacturing capabilities, and if we fail to develop and maintain supply relationships with various third-party manufacturers, we may be unable to develop or commercialize our drug
candidates.
|
|
|
• |
The design, development, manufacture, supply, and distribution of our drug candidates are highly regulated and technically complex.
|
|
|
• |
The other risks and uncertainties identified in Item 1A. Risk Factors and Item 7 Management’s Discussion and Analysis of Financial Condition and Results of Operations included in our Annual Report on Form
10-K for the year ended March 31, 2024.
|
All of the forward-looking statements in this quarterly report are qualified by this cautionary statement. There can be no guarantee that the results or developments that we anticipate will be
realized or, even if substantially realized, that they will have the consequences or effects on our business, financial condition, or results of operations that we anticipate. As a result, you should not place undue reliance on these
forward-looking statements. Except as required by applicable law, we do not undertake to update or amend any forward-looking statements, whether as a result of new information, future events or otherwise. All forward-looking statements are made
as of the date of this quarterly report.
We express all amounts in this quarterly report in thousands of U.S. dollars, except share and per share amounts or otherwise indicated. References to “$” are to U.S. dollars and references to
“CAD$” are to Canadian dollars.
Except as otherwise indicated, references in this quarterly report to “Grace,” “Grace Therapeutics”, “Acasti,” “the Company,” “we,” “us” and “our” refer to Grace Therapeutics, Inc. (formerly
known as Acasti Pharma Inc.) and its consolidated subsidiary.
PART I. FINANCIAL INFORMATION
Unaudited Condensed Consolidated Financial Statements
|
6
|
|
|
7
|
|
|
8
|
|
|
9
|
|
|
10
|
GRACE THERAPEUTICS, INC.
(Unaudited)
|
September 30,
2024
|
March 31,
2024
|
|||||||
|
(Expressed in thousands except share data)
|
|
$ |
|
$ |
||||
|
Assets
|
||||||||
|
Current assets:
|
||||||||
|
Cash and cash equivalents
|
|
|
||||||
|
Short-term investments
|
|
|
||||||
|
Receivables
|
|
|
||||||
|
Prepaid expenses
|
|
|
||||||
|
Total current assets
|
|
|
||||||
|
Equipment, net
|
|
|
||||||
|
Intangible assets
|
|
|
||||||
|
Goodwill
|
|
|
||||||
|
Total assets
|
|
|
||||||
|
Liabilities and shareholders’ equity
|
||||||||
|
Current liabilities:
|
||||||||
|
Trade and other payables
|
|
|
||||||
|
Total current liabilities
|
|
|
||||||
|
Derivative warrant liabilities
|
|
|
||||||
|
Deferred tax liability
|
|
|
||||||
|
Total liabilities
|
|
|
||||||
|
Commitments and contingencies (Note 11)
|
||||||||
|
Shareholders’ equity:
|
||||||||
|
Class A common shares,
|
|
|
||||||
|
Class B, C, D and E common shares,
|
|
|
||||||
|
Additional paid-in capital
|
|
|
||||||
|
Accumulated other comprehensive loss
|
(
|
)
|
(
|
)
|
||||
|
Accumulated deficit
|
(
|
)
|
(
|
)
|
||||
|
Total shareholders’ equity
|
|
|
||||||
|
Total liabilities and shareholders’ equity
|
|
|
||||||
See accompanying notes to unaudited condensed consolidated financial statements.
GRACE THERAPEUTICS, INC.
(Unaudited)
|
Three months ended
|
Six months ended
|
|||||||||||||||
|
September 30,
2024
|
September 30,
2023
|
September 30,
2024
|
September 30,
2023
|
|||||||||||||
|
(Expressed in thousands, except share and per share data)
|
|
$ |
|
$ |
|
$ |
|
$ |
||||||||
|
Operating expenses
|
||||||||||||||||
|
Research and development expenses, net of government assistance
|
(
|
)
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||||
|
General and administrative
|
(
|
)
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||||
|
Restructuring cost
|
|
|
|
(
|
)
|
|||||||||||
|
Loss from operating activities
|
(
|
)
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||||
|
Foreign exchange gain (loss)
|
|
(
|
)
|
|
(
|
)
|
||||||||||
|
Change in fair value of derivative warrant liabilities
|
|
(
|
)
|
|
(
|
)
|
||||||||||
|
Interest and other income, net
|
|
|
|
|
||||||||||||
|
Total other income (expenses), net
|
|
(
|
)
|
|
(
|
)
|
||||||||||
|
Loss before income tax benefit
|
(
|
)
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||||
|
Income tax benefit
|
|
|
|
|
||||||||||||
|
Net loss and total comprehensive loss
|
(
|
)
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||||
|
Basic and diluted loss per share
|
(
|
)
|
(
|
)
|
(
|
)
|
(
|
)
|
||||||||
|
Weighted average number of shares outstanding
|
|
|
|
|
||||||||||||
See accompanying notes to unaudited condensed consolidated financial statements.
GRACE THERAPEUTICS, INC.
|
Class A common shares
|
||||||||||||||||||||||||
|
(Expressed in thousands except share data)
|
Number
|
Amount
|
Additional paid-
in
capital
|
Accumulated
other
comprehensive
loss
|
Accumulated
deficit
|
Total
shareholders’
equity
|
||||||||||||||||||
|
|
$ |
|
$ |
|
$ |
|
$ |
|
$ |
|||||||||||||||
|
Balance, March 31, 2024
|
|
|
|
(
|
)
|
(
|
)
|
|
||||||||||||||||
|
Issuance of common shares upon cashless exercise of pre-funded warrants
|
|
|
|
|
|
|
||||||||||||||||||
|
Net loss and total comprehensive loss for the period
|
—
|
|
|
|
(
|
)
|
(
|
)
|
||||||||||||||||
|
Stock-based compensation
|
—
|
|
|
|
|
|
||||||||||||||||||
|
Balance at June 30, 2024
|
|
|
|
(
|
)
|
(
|
)
|
|
||||||||||||||||
|
Net loss and total comprehensive loss for the period
|
|
|
|
(
|
)
|
(
|
)
|
|||||||||||||||||
|
Stock-based compensation
|
—
|
|
|
|
|
|
||||||||||||||||||
|
Balance at September 30, 2024
|
|
|
|
(
|
)
|
(
|
)
|
|
||||||||||||||||
|
Class A common shares
|
||||||||||||||||||||||||
|
(Expressed in thousands except share data)
|
Number
|
Amount
|
Additional paid-
in
capital
|
Accumulated
other
comprehensive
loss
|
Accumulated
deficit
|
Total shareholders’
equity
|
||||||||||||||||||
|
|
$ |
|
$ |
|
$ |
|
$ |
|
$ |
|||||||||||||||
|
Balance, March 31, 2023
|
|
|
|
(
|
)
|
(
|
)
|
|
||||||||||||||||
|
Net loss and total comprehensive loss for the period
|
—
|
|
|
|
( |
) |
(
|
)
|
||||||||||||||||
|
Stock-based compensation
|
—
|
|
|
|
|
|
||||||||||||||||||
|
Balance at June 30, 2023
|
|
|
|
(
|
)
|
(
|
)
|
|
||||||||||||||||
|
Issuance of common shares and pre-funded warrants through private placement, net of offering costs
|
|
|
|
|
|
|
||||||||||||||||||
|
Issuance of common shares upon the exercise of stock options
|
|
|
|
|
|
|
||||||||||||||||||
|
Net loss and total comprehensive loss for the period
|
—
|
|
|
|
(
|
)
|
(
|
)
|
||||||||||||||||
|
Stock-based compensation
|
—
|
|
|
|
|
|
||||||||||||||||||
|
Balance at September 30, 2023
|
|
|
|
(
|
)
|
(
|
)
|
|
||||||||||||||||
See accompanying notes to unaudited condensed consolidated financial statements.
GRACE THERAPEUTICS, INC.
(Unaudited)
|
Six months ended
|
||||||||
|
September 30,
2024
|
September 30,
2023
|
|||||||
|
(Expressed in thousands)
|
|
$ |
|
$ |
||||
|
Cash flows used in operating activities:
|
||||||||
|
Net loss
|
(
|
)
|
(
|
)
|
||||
|
Adjustments:
|
||||||||
|
Depreciation expense
|
|
|
||||||
|
Gain on sale of equipment
|
|
(
|
)
|
|||||
|
Stock-based compensation
|
|
|
||||||
|
Change in fair value of derivative warrant liabilities
|
(
|
)
|
|
|||||
|
Deferred income tax benefit
|
(
|
)
|
(
|
)
|
||||
|
Changes in operating assets and liabilities:
|
||||||||
|
Receivables
|
|
(
|
)
|
|||||
|
Prepaid expenses
|
(
|
)
|
(
|
)
|
||||
|
Trade and other payables
|
|
(
|
)
|
|||||
|
Operating lease right of use asset
|
|
(
|
)
|
|||||
|
Net cash used in operating activities
|
(
|
)
|
(
|
)
|
||||
|
Cash flows from investing activities:
|
||||||||
|
Proceeds from sale of equipment
|
|
|
||||||
|
Purchase of short-term investments
|
(
|
)
|
|
|||||
|
Net cash (used in) provided by investing activities
|
(
|
)
|
|
|||||
|
Cash flows from financing activities:
|
||||||||
|
Net proceeds from issuance of common shares and warrants from private Placement
|
|
|
||||||
|
Proceeds from issuance of common shares from exercise of stock options
|
|
|
||||||
|
Net cash provided by financing activities
|
|
|
||||||
|
Net decrease in cash and cash equivalents
|
(
|
)
|
(
|
)
|
||||
|
Cash and cash equivalents, beginning of period
|
|
|
||||||
|
Cash and cash equivalents, end of period
|
|
|
||||||
|
Cash and cash equivalents are comprised of:
|
||||||||
|
Cash
|
|
|
||||||
|
Cash equivalents
|
|
|
||||||
See accompanying notes to unaudited condensed consolidated financial statements.
GRACE THERAPEUTICS, INC.
(Unaudited)
(Expressed in thousands except share and per share data)
1. Nature of operation
General
Grace Therapeutics, Inc. (formerly known as Acasti Pharma Inc.) (“Acasti Delaware” or “the Company”), is a Delaware corporation that, as further described below, previously existed under the laws of the
Province of Québec, Canada (“Acasti Québec”), before changing its jurisdiction on October 1, 2024 to the Province of British Columbia, Canada (“Acasti British Columbia”). On October 7, 2024, Acasti British Columbia changed its jurisdiction to
the State of Delaware. Effective October 28, 2024, the Company changed its corporate name to Grace Therapeutics, Inc.
Continuance and Domestication
On October 1, 2024, Acasti Québec changed its jurisdiction of incorporation from
the Province of Québec in Canada to the Province of British Columbia in Canada pursuant to a “continuance” effected in accordance with Chapter XII of the Business Corporations Act (Québec) (the “Continuance”). Subsequently on October 7, 2024,
Acasti British Columbia changed its jurisdiction of incorporation from the Province of British Columbia in Canada to the State of Delaware in the United States of America pursuant to a “continuance” effected in accordance with Section 308 of
the Business Corporations Act (British Columbia) and a “domestication” (the “Domestication”) under Section 388 of the General Corporation Law of the State of Delaware (the “DGCL). Both the Continuance and the Domestication were approved by the
Company’s shareholders at the Company’s Annual and Special Meeting of Shareholders held on September 30, 2024.
Prior to the Continuance and Domestication, the Company’s Class A common shares,
without par value per share (“Common Shares”), were listed on The Nasdaq Stock Market LLC (“Nasdaq”) under symbol “ACST.” Upon the effectiveness of the Continuance, each outstanding Class A common share of Acasti Québec at the time of the
Continuance remained issued and outstanding as a common share, without par value per share, of Acasti British Columbia. Upon effectiveness of the Domestication, each outstanding common share of Acasti British Columbia at the time of the
Domestication automatically became one outstanding share of common stock, par value $0.0001 per share, of Acasti Delaware. The Company’s common stock continues to be listed for trading
on Nasdaq and in connection with its corporate name change to Grace Therapeutics, Inc., commenced trading under the symbol “GRCE” on October 28, 2024.
The Continuance and Domestication
will be accounted for as an exchange of equity interest among entities under common control resulting in a change in reporting entity, and will be reflected when the financial statements are issued for a period that includes the date the
consummation of the Domestication, which is October 7, 2024 (the “Effective Date”). All assets and liabilities of Acasti British Columbia were assumed by the Company at the Effective Date, resulting in the retention of the historical basis of
accounting as if they had always been combined for accounting and financial reporting purposes. Any excess resulting from the automatic conversion of each of the outstanding common share of Acasti British Columbia into one outstanding share of
common stock of Acasti Delaware, will be presented as Additional Paid-in Capital in the equity section of the balance sheet.
Liquidity and Financial Condition
The Company has incurred operating losses and negative cash flows from operations in each period since its inception. The Company expects to incur significant expenses and continued operating losses for the foreseeable
future.
In May 2023, the Company implemented a strategic realignment plan to enhance
shareholder value that resulted in the Company engaging a new management team, streamlining its research and development activities, and greatly reducing its workforce. Following the realignment, the Company is a smaller, more focused
organization, based in the United States, and concentrated on its development of its lead product candidate GTx-104. Further development of GTx-102 and GTx-101 will occur at such a time when the Company is able to secure additional funding or
enters into strategic partnerships for license or sale with third parties.
On September 24, 2023, the Company entered into a securities purchase agreement with certain institutional and accredited investors. Gross
proceeds to the Company from this private placement were approximately $7.5 million, before deducting fees and expenses. The Company
issued and sold an aggregate of 1,951,371 Common Shares, pre-funded warrants (the “Pre-funded Warrants”) to purchase up to an
aggregate of 2,106,853 Common Shares, each at a purchase price of $1.848 per Common Share and accompanying common warrants (the “Common Warrants” and, together with the Pre-funded Warrants, the “Warrants”) to purchase up to an aggregate of 2,536,391 Common Shares. The Company currently uses the net proceeds from the private placement for clinical trial expenses to further the Phase 3
clinical trial for GTx-104, pre-commercial planning, working capital and other general corporate purposes. The Company believes its existing cash and cash equivalents will be sufficient to fund the Company’s operations into the second calendar
quarter of 2026.
The Company will require additional capital to fund its daily operating needs beyond that time. The Company does not expect to generate revenue from product sales unless and
until it successfully completes drug development and obtains regulatory approval, which the Company expects will take a few years and is subject to significant uncertainty. To date, the Company has financed its operations primarily through
public offerings and private placements of its Common Shares, warrants and convertible debt and the proceeds from research tax credits. Until such time that the Company can generate significant revenue from drug product sales, if ever, it will
require additional financing, which is expected to be sourced from a combination of public or private equity or debt financing or other non-dilutive sources, which may include fees, milestone payments and royalties from collaborations with
third parties. Arrangements with collaborators or others may require the Company to relinquish certain rights related to its technologies or drug product candidates. Adequate additional financing may not be available to the Company on
acceptable terms, or at all. The Company’s inability to raise capital as and when needed could have a negative impact on its financial condition and its ability to pursue its business strategy. The Company plans to raise additional capital in
order to maintain adequate liquidity. Negative results from studies or trials, if any, or depressed prices of the Company’s stock could impact the Company’s ability to raise additional financing. Raising additional equity capital is subject to
market conditions that are not within the Company’s control. If the Company is unable to raise additional funds, the Company may not be able to realize its assets and discharge its liabilities in the normal course of business.
The Company remains subject to risks similar to other development stage companies in the biopharmaceutical industry, including compliance with
government regulations, protection of proprietary technology, dependence on third-party contractors and consultants and potential product liability, among others. Please refer to the risk factors included in Part 1, Item 1A of the Company’s
Annual Report on Form 10-K for the year ended March 31, 2024, filed with the SEC on June 21, 2024 (the “Annual Report”).
2. Summary of significant accounting policies:
Basis of presentation
The accompanying unaudited condensed consolidated financial statements have been prepared in accordance with generally accepted accounting
principles in the United States of America (“U.S. GAAP”) for interim financial information and with the instructions to Form 10-Q and Article 8 of Regulation S-X under the Securities Exchange Act of 1934. Any reference in these notes to
applicable guidance is meant to refer to the authoritative U.S. GAAP as found in the Accounting Standards Codification (“ASC”) and as amended by Accounting Standards Updates (“ASU”) of the Financial Accounting Standards Board (“FASB”).
The unaudited condensed consolidated financial statements have been prepared on the same basis as the audited annual consolidated financial
statements as of and for the year ended March 31, 2024, and, in the opinion of management, reflect all adjustments, consisting of normal recurring adjustments, necessary for the fair presentation of the Company’s consolidated financial position
as of September 30, 2024, the consolidated results of its operations for the three and the six months ended September 30, 2024 and 2023, its statements of shareholders’ equity for the six months ended September 30, 2024 and 2023, and its
consolidated cash flows for the six months ended September 30, 2024 and 2023.
These unaudited condensed consolidated financial statements should be read in conjunction with the Company’s audited consolidated financial
statements and the accompanying notes for the year ended March 31, 2024 included in the Company’s Annual Report. The condensed consolidated balance sheet data as of March 31, 2024 presented for comparative purposes was derived from the Company’s
audited consolidated financial statements. The results for the three and the six months ended September 30, 2024 are not necessarily indicative of the operating results to be
expected for the full year or for any other subsequent interim period.
The Company’s significant accounting policies are disclosed in the audited consolidated financial statements for the year ended March 31, 2024
included in the Annual Report. There have been no changes to the Company’s significant accounting policies since the date of the audited consolidated financial statements for the year ended March 31, 2024 included in the Annual Report.
Use of estimates
The preparation of these unaudited condensed consolidated financial statements in conformity with U.S. GAAP requires management to make
estimates and assumptions that affect the reported amounts of assets, liabilities, income, and expenses. Actual results may differ from these estimates.
Estimates are based on management’s best knowledge of current events and actions that management may undertake in the future. Estimates and
underlying assumptions are reviewed on an ongoing basis. Revisions to accounting estimates are recognized in the period in which the estimates are revised and in any future periods affected.
Estimates and assumptions include the measurement of stock-based compensation, derivative warrant liabilities, accruals for research and
development contracts and contract organization agreements, and valuation of intangibles and goodwill. Estimates and assumptions are also involved in determining the extent to which research and development expenses qualify for research and
development tax credits. The Company recognizes tax credits once it has reasonable assurance that they will be realized.
Reclassifications
Certain reclassifications have been made to prior period amounts to conform to current period reporting classifications.
Recent accounting pronouncements
The Company has considered recent accounting pronouncements and concluded that they are either not applicable to the Company’s business or that
the effect is not expected to be material to the unaudited condensed consolidated financial statements as a result of future adoption.
3. Fair Value Measurements
Assets and liabilities measured at fair value on a recurring basis as of September 30, 2024 are as follows:
|
Total
|
Quoted prices in
active markets
(Level 1)
|
Significant other
observable
inputs (Level 2)
|
Significant
unobservable
inputs (Level 3)
|
|||||||||||||
|
$
|
$
|
$
|
$
|
|||||||||||||
|
Assets
|
||||||||||||||||
|
Treasury bills and term deposits classified as cash equivalents
|
|
|
|
|
||||||||||||
|
Guaranteed investment certificate classified as short-term investments
|
|
|
|
|
||||||||||||
|
Total assets
|
|
|
|
|
||||||||||||
|
Liabilities
|
||||||||||||||||
|
Derivative warrant liabilities
|
|
|
|
|
||||||||||||
|
Total liabilities
|
|
|
|
|
||||||||||||
Assets and liabilities measured at fair value on a recurring basis as of March 31, 2024 are as follows:
|
Total
|
Quoted prices
in active
markets
(Level 1)
|
Significant other
observable
inputs (Level 2)
|
Significant
unobservable
inputs (Level 3)
|
|||||||||||||
|
|
$ |
|
$ |
|
$ |
|
$ | |||||||||
|
Assets
|
||||||||||||||||
|
Guaranteed investment certificates and term deposits classified as cash equivalents
|
|
|
|
|
||||||||||||
|
Total assets
|
|
|
|
|
||||||||||||
|
Liabilities
|
||||||||||||||||
|
Derivative warrant liabilities
|
|
|
|
|
||||||||||||
|
Total liabilities
|
|
|
|
|
||||||||||||
There were no changes in valuation techniques or transfers between Levels 1, 2 or 3 during the three and the six months ended September 30,
2024. The Company’s derivative warrant liabilities are measured at fair value on a recurring basis using unobservable inputs that are classified as Level 3 inputs. Refer to Note 8 for the valuation techniques and assumptions used in estimating
the fair value of the derivative warrant liabilities.
4. Receivables
|
September 30,
2024
|
March 31,
2024
|
|||||||
|
|
$ |
|
$ | |||||
|
Sales tax receivables
|
|
|
||||||
|
Government assistance
|
|
|
||||||
|
Interest receivable
|
|
|
||||||
|
Other receivables
|
|
|
||||||
|
Total receivables
|
|
|
||||||
Government assistance
is comprised of research and development investment tax credits from the Québec provincial government, which relate to quantifiable research and development expenditures under the applicable tax laws. The amounts recorded as receivable are
subject to a government tax audit and the final amounts received may differ from those recorded.
5. Short-term investments
The Company holds various marketable securities, with maturities greater than 90 days at the time of purchase, as follows:
|
September 30,
2024
|
March 31,
2024
|
|||||||
|
|
$ |
|
$ | |||||
|
Guaranteed investment certificate issued in CAD currency earning interest at
|
|
|
||||||
|
Total short-term investments
|
|
|
||||||
6. Trade and other
payables
|
September 30,
2024
|
March 31,
2024
|
|||||||
|
|
$ |
|
$ | |||||
|
Trade payables
|
|
|
||||||
|
Accrued liabilities and other payables
|
|
|
||||||
|
Employee salaries and benefits payable
|
|
|
||||||
|
Total trade and other payables
|
|
|
||||||
7. Leases
The Company has historically entered lease arrangements for its research and
development and quality control laboratory facility located in Sherbrooke, Québec. In March 2022, the Company renewed the lease agreement effective April 1, 2022, resulting in a commitment of $556 over a 24 -month base lease term with an option to renew
for an additional 48 -month term. In April 2023, the Company elected not to renew the additional 48-month option to renew and
terminated the lease on March 31, 2024. As of September 30, 2024, the Company had one month to month lease for its principal executive
offices in Princeton Junction, NJ.
Lease expense related to leases is as follows:
|
Three months ended
|
Six months ended
|
|||||||||||||||
|
September 30,
2024
|
September 30,
2023
|
September 30,
2024
|
September 30,
2023
|
|||||||||||||
|
|
$ |
|
$ |
|
$ |
|
$ | |||||||||
|
Operating lease cost
|
|
|
|
|
||||||||||||
|
Total lease expense
|
|
|
|
|
||||||||||||
8. Shareholders’ Equity
Private Placement
In September 2023, the Company entered into a securities purchase agreement (the
“Purchase Agreement”) with certain institutional and accredited investors in connection with a private placement of the Company’s securities (the “Offering”). Pursuant to the Purchase Agreement, the Company agreed to offer and sell 1,951,371 Common Shares, at a purchase price of $1.848
per Common Share and Pre-funded Warrants to purchase up to 2,106,853 Common Shares at a purchase price equal to the purchase price per
Common Share less $0.0001 . Each Pre-funded Warrant is exercisable for one Common Share at an exercise price of $0.0001 per Common Share, is
immediately exercisable, and will expire once exercised in full. Pursuant to the Purchase Agreement, the Company also issued, to such institutional and accredited investors, Common Warrants to purchase Common Shares exercisable for an aggregate
of 2,536,391 Common Shares. Under the terms of the Purchase Agreement, for each Common Share and each Pre-funded Warrant issued in the
Offering, an accompanying five-eighths (0.625 ) of a Common Warrant was issued to the purchaser thereof. Each whole Common Warrant is
exercisable for one Common Share at an exercise price of $3.003 per Common Share, is immediately exercisable, and will expire on the earlier of (i) the 60th day after the date of the acceptance by the FDA of an NDA for the Company’s
product candidate GTx-104 or (ii) five years from the date of issuance. The Offering closed on September 25, 2023. The net proceeds
to the Company from the Offering were $7,338 , after deducting fees and expenses.
The Offering included the issuance of Common Shares, Pre-funded Warrants, and Common Warrants to related parties Shore Pharma LLC, an entity
that was controlled by Vimal Kavuru, the Chair of our Board of Directors, at the time of the Offering and SS Pharma LLC, the beneficial owner of 5.5 %
of Common Stock outstanding prior to the Offering, resulting in proceeds of $2,500 . As of September 30, 2024 and March 31, 2024, the
balance of derivative warrant liabilities from these related parties was $868 and $1,453 , respectively.
During the six months ended September 30, 2024, of the 2,106,853 Pre-funded Warrants, 740,480
were exercised into Common Shares.
Warrants
As further discussed above, on September 25, 2023, the Company issued Pre-Funded
Warrants and Common Warrants exercisable for an aggregate of 4,643,244 Common Shares in the Offering pursuant to the terms of the
Purchase Agreement entered into with certain institutional and accredited investors.
The Common Warrants issued as a part of the Offering are derivative warrant
liabilities given the Common Warrants did not meet the fixed-for-fixed criteria and that the Common Warrants are not indexed to the Company’s own stock. Proceeds were allocated amongst Common Shares, Pre-funded Warrants, and Common Warrants by
applying the residual method, with fair value of the Common Warrants determined using the Black-Scholes model, resulting in initial derivative warrant liabilities of $1,631 and issuance costs of $45 allocated to Common Warrants. Accordingly, $2,822 and $3,047 of the gross proceeds
were allocated to Common Shares and Pre-funded Warrants, respectively, and $78 and $84 of issuance costs were allocated to Common Shares and Pre-funded Warrants, respectively.
The derivative warrant liabilities are measured at fair value at each reporting
period and the reconciliation of changes in fair value is presented in the following table:
|
September 30,
2024
|
September 30,
2023
|
|||||||
|
|
$ |
|
$ | |||||
|
Beginning balance
|
|
|
||||||
|
Issued during the period
|
|
|
||||||
|
Change in fair value
|
(
|
)
|
|
|||||
|
Ending balance
|
|
|
||||||
The fair
value of derivative warrant liabilities were determined based on the fair value of the Common Warrants at the issue date and the reporting dates using the Black-Scholes model with the following weighted-average assumptions that will expire on
the earlier of (i) the 60th day after the date of the acceptance by the FDA of an NDA for the Company’s product candidate GTx-104 or (ii) five years from the date on issuance.
|
September 30,
2024
|
March 31,
2024
|
|||||||
|
Risk-free interest rate
|
|
%
|
|
%
|
||||
|
Share price
|
$
|
|
$
|
|
||||
|
Expected warrant life
|
|
|
||||||
|
Dividend yield
|
|
%
|
|
%
|
||||
|
Expected volatility
|
|
%
|
|
%
|
||||
The
weighted-average fair values of the Common Warrants were determined to be $1.03 and $1.72 per Common Warrant as of September 30, 2024, and March 31, 2024, respectively. The risk-free interest rate at the issue date and on the reporting date of September 30,
2024 was based on the interest rate corresponding to the U.S. Treasury rate issue with a remaining term equal to the expected term of the Common Warrants. The expected volatility was based on the historical volatility for the Company.
At September 30, 2024, the Company had outstanding Common Warrants
to purchase 2,536,391 Common Shares, with an exercise price of $3.003 , all of which were classified as derivative warrant liabilities. During the six months ended September 2024, 740,480 Pre-funded Warrants were exercised into 740,457
Common Shares. At September 30, 2024, the Company had outstanding Pre-funded Warrants to purchase 1,366,373 Common Shares, with an
exercise price of $0.0001 , all of which were classified within shareholders’ equity.
In connection with the Continuance and the Domestication, the Company continues
its obligations under the Purchase Agreement and the related Pre-Funded and Common Stock Warrants. Upon effectiveness of the Continuance, each outstanding warrant settleable into Class A common shares of Acasti Québec remained exercisable for or
able to be settled into an equivalent number of common shares of Acasti British Columbia for the equivalent exercise price per share (if applicable), without any action by the holder. Upon effectiveness of the Domestication, each outstanding
warrant settleable into common shares of Acasti British Columbia remained exercisable for or able to be settled into an equivalent number of shares of common stock of Acasti Delaware for the equivalent exercise price per share (if applicable).
9. Stock-based compensation
Stock option plan
At September 30,
2024, the Company had in place a stock option plan for directors, officers, employees, and consultants of the Company (“Stock Option Plan”). As of September 30, 2024, there were 540,595 awards available under the Stock Option Plan for issuance.
The Stock Option Plan
provides for the granting of options to purchase Common Shares. Under the terms of the Stock Option Plan, the exercise price of the stock options granted under the Stock Option Plan may not be lower than the closing price of the Company’s Common
Shares on the Nasdaq Capital Market at the close of such market the day preceding the grant. The maximum number of Common Shares that may be issued upon exercise of options granted under the Stock Option Plan shall not exceed 20 % of the aggregate number of issued and outstanding shares of the Company as of July 28, 2022. The terms and conditions for acquiring and exercising
options are set by the Company’s Board of Directors, subject to, among others, the following limitations: the term of the options cannot exceed ten years and (i) all options granted to a director will be vested evenly on a monthly basis over a period of at least twelve (12) months, and (ii) all options granted to an employee will be vested evenly on a quarterly basis over
a period of at least thirty-six (36) months.
The total number of
options issued to any one consultant within any twelve-month period cannot exceed 2 % of the Company’s total issued and outstanding
Common Shares (on a non-diluted basis). The total number of options issued within any twelve-month period to all directors, employees and/or consultants of the Company (or any subsidiary of the Company) conducting investor relations services
cannot exceed in the aggregate 2 % of the Company’s issued and outstanding Common Shares (on a non-diluted basis), calculated at the
date an option is granted to any such person.
The following table summarizes information about activities within the Stock
Option Plan for the six months period ended September 30, 2024:
|
Number of
options
|
Weighted-average
exercise price
|
Remaining
Contractual Term
(years)
|
Aggregate Intrinsic
Value (in
thousands)
|
|||||||||||||
| $ |
|
$ |
||||||||||||||
|
Outstanding, March 31, 2024
|
|
|
|
|
||||||||||||
|
Granted
|
|
|
—
|
—
|
||||||||||||
|
Outstanding, September 30, 2024
|
|
|
|
|
||||||||||||
|
Exercisable, September 30, 2024
|
|
|
|
|
||||||||||||
The weighted-average grant date fair value of awards for options
granted during the six months ended September 30, 2024 was $2.52 . The fair value of options granted was estimated using the
Black-Scholes option pricing model, resulting in the following weighted-average assumptions for the options granted:
|
September 30, 2024
|
September 30, 2023
|
|||||||
|
Weighted-average
|
Weighted-average
|
|||||||
|
Exercise price
|
$
|
|
$
|
|
||||
|
Share price
|
$
|
|
$
|
|
||||
|
Dividend
|
|
|
||||||
|
Risk-free interest
|
|
%
|
|
%
|
||||
|
Estimated life (years)
|
|
|
||||||
|
Expected volatility
|
|
%
|
|
%
|
||||
Compensation expense recognized under the Stock Option Plan is summarized as
follows:
|
Three months ended
|
Six months ended
|
|||||||||||||||
|
September 30,
2024
|
September 30,
2023
|
September 30,
2024
|
September 30,
2023
|
|||||||||||||
|
|
$ |
|
$ |
|
$ |
|
$ | |||||||||
|
Research and development expenses
|
|
|
|
|
||||||||||||
|
General and administrative expenses
|
|
|
|
|
||||||||||||
|
|
|
|
|
|||||||||||||
As of September 30, 2024, there was $513 of total unrecognized compensation cost, related to non-vested stock options, which is expected to be recognized over a remaining
weighted-average vesting period of 1.31 years.
Equity incentive plan
The Company established an equity incentive plan (the “Equity Incentive Plan”)
for employees, directors, and consultants. The Equity Incentive Plan provides for the issuance of 1,483,140 restricted share units,
performance share units, restricted shares, deferred share units and other stock-based awards, subject to restricted conditions as may be determined by the Board of Directors. There were no such awards outstanding at September 30, 2024, and no
In connection with the Continuance and the Domestication, the Company continues
its obligations under (i) the Stock Option Plan and (ii) the Equity Incentive Plan (together, the “Prior Plans”) and all of the outstanding equity awards under the Prior Plans. Upon effectiveness of the Continuance, each outstanding option,
equity awards and restricted share unit settleable into Class A common shares of Acasti Québec remained exercisable for or able to be settled into an equivalent number of common shares of Acasti British Columbia for the equivalent exercise price
per share (if applicable), without any action by the holder. Upon effectiveness of the Domestication, each outstanding option, equity awards and restricted share unit settleable into common shares of Acasti British Columbia remained exercisable
for or able to be settled into an equivalent number of shares of common stock of Acasti Delaware for the equivalent exercise price per share (if applicable), without any action by the holder.
Following the Effective Date of the 2024 Plan (each as defined in Note 13), no awards shall be made under the Prior Plans. However, shares of
common stock reserved under the Prior Plans to settle awards which are made under the Prior Plans prior to the Effective Date may be issued and delivered following the Effective Date to settle such awards.
10. Loss per share
The Company has generated a net loss for all periods presented. Therefore,
diluted loss per share is the same as basic loss per share since the inclusion of potentially dilutive securities would have had an anti-dilutive effect. All currently outstanding options and warrants could potentially be dilutive in the future.
The Company excluded the following potential Common Shares, presented based on
amounts outstanding at each period end, from the computation of diluted net loss per share attributable to common shareholders for the periods indicated because including them would have had an anti-dilutive effect:
|
Six months ended
|
||||||||
|
September 30, 2024
|
September 30, 2023
|
|||||||
|
Options outstanding
|
|
|
||||||
|
September 2023 Common Warrants
|
|
|
||||||
Basic and diluted net loss per share is calculated based upon the
weighted-average number of Common Shares outstanding during the period. Common Shares underlying the Pre-funded Warrants are included in the calculation of basic and diluted earnings per share.
11. Commitments and
contingencies
Research and
development contracts and contract research organizations agreements
The Company utilizes contract manufacturing organizations (“CMOs”) for the development and production of clinical materials and contract research organizations (“CROs”) to perform services related to its clinical trials. Pursuant to the agreements with these CMOs and CROs, the Company has either the right to terminate the agreements without
penalties or under certain penalty conditions. As of September 30, 2024, the Company has $398 of commitments to CMOs and $3.9 million of commitments to CROs for the next twelve months.
Raw krill oil supply
contract
On October 25, 2019, the Company signed a
supply agreement with Aker BioMarine Antarctic AS. (“AKBM”) to purchase raw krill oil product for a committed volume of commercial starting material for CaPre, one of the Company’s former drug candidates, for a total fixed value of $3.1 million based on the value of krill oil at that time. As of March 31, 2022, the remaining balance of commitment amounted to $2.8 million. During the second calendar quarter of 2022, AKBM informed the Company that AKBM believed it had satisfied the terms of the supply
agreement as to their obligation to deliver the remaining balance of raw krill oil product, and that the Company was therefore required to accept the remaining product commitment. The Company disagreed with AKBM’s position and believed that AKBM
was not entitled to further payment under the supply agreement. Accordingly, no liability was recorded by the Company. The dispute remained unresolved as of both March 31, 2023 and 2022. On October 18, 2023, the Company entered into an agreement
with AKBM to settle any and all potential claims regarding amounts due under the supply agreement (“Settlement Agreement”). Pursuant to the terms of the Settlement Agreement, in exchange for a release and waiver of claims arising out of the
supply agreement by AKBM and any of AKBM’s affiliates, the Company and AKBM agreed to the following: (a) AKBM retained ownership of all raw krill oil product, including amounts previously delivered to the Company, (b) AKBM acquired and took
ownership of all production equipment related to the production of CaPre, (c) AKBM acquired and took ownership of all data from research, clinical trials and pre-clinical studies with respect to CaPre, and (d) AKBM acquired and took ownership
over all rights, title and interest in and to all intellectual property rights, including all patents and trademarks, related to CaPre owned by the Company. Pursuant to the terms of the Settlement Agreement, AKBM acknowledged that the CaPre
assets were transferred on an “as is” basis, and in connection therewith the Company disclaimed all representations and warranties in connection with the CaPre assets, including any representations with respect to performance or sufficiency. The
value of the raw krill oil previously delivered to the Company, the production equipment, and the intellectual property rights related to CaPre were fully impaired in prior reporting periods and had a carrying value of zero as of March 31, 2023.
For the three and the six months ended September 30, 2024, there was and $193 , respectively, in expenses recorded by the Company in relation to shipping cost to transport the Company’s production equipment related to the production of CaPre.
Legal proceedings and
disputes
In the ordinary course of
business, the Company is at times subject to various legal proceedings and disputes. The Company assesses its liabilities and contingencies in connection with outstanding legal proceedings utilizing the latest information available. Where it is
probable that the Company will incur a loss and the amount of the loss can be reasonably estimated, the Company records a liability in its consolidated financial statements. These legal contingencies may be adjusted to reflect any relevant
developments. Where a loss is not probable or the amount of loss is not estimable, the Company does not accrue legal contingencies. While the outcome of legal proceedings is inherently uncertain, based on information currently available,
management believes that it has established appropriate legal reserves. Any incremental liabilities arising from pending legal proceedings are not expected to have a material adverse effect on the Company’s financial position, results of
operations, or cash flows. However, it is possible that the ultimate resolution of these matters, if unfavorable, may be material to the Company’s financial position, results of operations, or cash flows. No reserves or liabilities have been accrued at September 30, 2024.
12. Restructuring costs
On May 8, 2023, the Company communicated
its decision to terminate a substantial amount of its workforce as part of a plan that intended to align the Company’s organizational and management cost structure to prioritize resources to GTx-104, thereby reducing losses to improve cash flow
and extend available cash resources. During the six months ended, September 30, 2023, the Company incurred $1,485 in costs primarily
consisting of employee severance costs and legal fees. There were no
13. Subsequent Events
| a. |
Adoption of
Delaware Certificate of Incorporation and Bylaws
|
In connection with
the consummation of the Domestication, on October 7, 2024, the Company adopted a Certificate of Incorporation (the “Charter”) and Bylaws (the “Bylaws”). The rights of holders of the Company’s common stock are now governed by the Charter, the
Bylaws, and the General Corporation Law of the State of Delaware.
| b. |
Indemnification Agreements
|
In connection with the consummation of the Domestication and pursuant to the Company’s Charter, Bylaws and the DGCL, on October 7, 2024, the Company entered into indemnification agreements with each of the
Company’s executive officers and directors providing for the indemnification of, and advancement of expenses to, each such person in connection with claims, suits or proceedings arising as a result of such person’s service as an officer or
director of the Company.
| c. |
2024 Equity Incentive Plan
|
At the Annual and
Special Meeting of Shareholders on September 30, 2024, the Company’s stockholders approved the Acasti Pharma Inc 2024 Equity Incentive Plan (the “2024 Plan”) which became effective on the date of the Domestication. The 2024 Plan provides for
the grant of awards of stock options, stock appreciation rights, restricted stock, restricted stock units, deferred stock units, unrestricted stock, dividend equivalent rights, performance-based awards and other equity-based awards to eligible
persons as defined under the 2024 Plan. Any of these awards may, but need not, be made as performance incentives to reward the holders of such awards for the achievement of performance goals in accordance with the terms of the 2024 Plan. Stock
options granted under the 2024 Plan may be non-qualified stock options or incentive stock options, as provided in the 2024 Plan.
The 2024 Plan is administered by a
committee designated from time to time, by resolution of the Company’s Board of Directors. The committee will also be responsible for determining, among others, the key terms of the awards including their grant dates, pricing, basis for fair
value determination, vesting terms, restrictions, and terminations. There are 1,350,000 shares of common stock available for issuance
under the 2024 Plan. No awards may be made under the Prior Plans. However, shares of common stock reserved under the Prior Plans to settle awards that were made under the Prior Plans prior to the Effective Date may be issued and delivered
following the Effective Date to settle such awards.
The 2024 Plan will terminate automatically
ten years after the Effective Date and maybe terminated on any earlier date as provided by the 2024 Plan, provided the incentive stock
options may not be granted under the 10th year anniversary of the Board’s adoption of the 2024 Plan.
This management’s discussion and analysis (“MD&A”) is presented in order to provide the reader with an overview of the financial results and
changes to our consolidated balance sheet at September 30, 2024. This MD&A also explains the material variations in our results of operations for the three and the six months ended September 30, 2024 and 2023, consolidated balance sheets as
of September 30, 2024 and March 31, 2024, and cash flows for the six months ended September 30, 2024 and 2023.
Market data, and certain industry data and forecasts included in this MD&A were obtained from internal Company surveys and market research conducted by third parties hired by us, publicly
available information, reports of governmental agencies and industry publications, and independent third-party surveys. We have relied upon industry publications as our primary sources for third-party industry data and forecasts. Industry
surveys, publications and forecasts generally state that the information they contain has been obtained from sources believed to be reliable, but that the accuracy and completeness of that information are not guaranteed. We have not independently
verified any of the data from third-party sources or the underlying economic assumptions they have made. Similarly, internal surveys, industry forecasts and market research, which we believe to be reliable based upon our management’s or
contracted third parties’ knowledge of our industry, have not been independently verified. Our estimates involve risks and uncertainties, including assumptions that may prove not to be accurate, and these estimates and certain industry data are
subject to change based on various factors, including those discussed in this quarterly report and in our most recently filed Annual Report on Form 10-K, filed with the Securities and Exchange Commission (the “SEC”) on June 21, 2024 (the “Annual
Report”). This MD&A contains forward-looking information. You should review our Special Note Regarding Forward-Looking Statements presented at the beginning of this quarterly report.
This MD&A should be read in conjunction with our unaudited condensed consolidated interim financial statements for the three and the six months ended September 30, 2024 and 2023 included
elsewhere in this quarterly report. Our unaudited condensed consolidated financial statements were prepared in accordance with U.S. GAAP.
All amounts appearing in this MD&A for the period-by-period discussions are in thousands of U.S. dollars, except share and per share amounts or unless otherwise indicated.
Business Overview
We are focused on developing and commercializing products for rare and orphan diseases that have the potential to improve clinical outcomes by
using our novel drug delivery technologies. We seek to apply new proprietary formulations to approved and marketed pharmaceutical compounds to achieve enhanced efficacy, faster onset of action, reduced side effects, more convenient drug
delivery and increased patient compliance; all of which could result in improved patient outcomes. The active pharmaceutical ingredients used in the drug candidates under development by Grace Therapeutics may be already approved in a target
indication or could be repurposed for use in new indications.
The existing well understood efficacy and safety profiles of these marketed compounds provide the opportunity for us to utilize the Section 505(b)(2) regulatory pathway under the Federal Food,
Drug and Cosmetic Act (“FDCA”) for the development of our reformulated versions of these drugs, and therefore may provide a potentially shorter path to regulatory approval. Under Section 505(b)(2), if sufficient support of a product’s safety and
efficacy either through previous U.S. Food and Drug Administration (“FDA”) experience or sufficiently within the existing and accepted scientific literature, can be established, it may eliminate the need to conduct some of the pre-clinical
studies and clinical trials that new drug candidates might otherwise require.
Our therapeutic pipeline consists of three unique clinical-stage drug candidates supported by an intellectual property portfolio of more than 40 granted and pending patents in various
jurisdictions worldwide. These drug candidates aim to improve clinical outcomes in the treatment of rare and orphan diseases by applying proprietary formulation and drug delivery technologies to existing pharmaceutical compounds to achieve
improvements over the current standard of care, or to provide treatment for diseases with no currently approved therapies.
We believe that rare disorders represent an attractive area for drug development, and there remains an opportunity for us to utilize already approved drugs that have established safety profiles and clinical experience
to potentially address significant unmet medical needs. A key advantage of pursuing therapies for rare disorders is the potential to receive orphan drug designation (“ODD”) from the FDA. Our three drug candidates have received ODD status,
provided certain conditions are met at new drug application (“NDA”) approval. ODD provides for seven years of marketing exclusivity in the United States post-launch, provided certain conditions are met, and the potential for faster regulatory
review. ODD status can also result in tax credits of up to 50% of clinical development costs conducted in the United States upon marketing approval and a waiver of the NDA fees, which we estimate can translate into savings of approximately $3.2
million for our lead drug candidate, GTx-104. Developing drugs for rare diseases can often allow for clinical trials that are more manageably scaled and may require a smaller, more targeted commercial infrastructure.
The specific diseases targeted for drug development by us are well understood, although the patient populations suffering from such diseases may remain poorly served by available therapies or,
in some cases, approved therapies do not yet exist. We aim to effectively treat debilitating symptoms that result from these underlying diseases.
Our management team possesses significant experience in drug formulation and drug delivery research and development, clinical and pharmaceutical
development and manufacturing, regulatory affairs, and business development, as well as being well-versed in late-stage drug development and commercialization. Importantly, our team is comprised of industry professionals with deep expertise and
knowledge, including a world-renowned practicing neurosurgeon-scientist and respected authority in aSAH, as well as product development, chemistry, manufacturing and controls (CMC”), planning, implementation, management, and execution of global
Phase 2 and Phase 3 trials for GTx-104, and drug commercialization.
Our Pipeline
|
|
• |
GTx-104 is a clinical-stage, novel, injectable formulation of nimodipine being developed for intravenous (“IV”) infusion in aneurysmal subarachnoid
hemorrhage (“aSAH”) patients to address significant unmet medical needs. The unique nanoparticle technology of GTx-104 facilitates aqueous formulation of insoluble nimodipine for a standard peripheral IV infusion. GTx-104 provides a
convenient IV delivery of nimodipine in the intensive care unit eliminating the need for nasogastric tube administration in unconscious or dysphagic patients. IV delivery of GTx-104 also has the potential to lower food effects,
drug-to-drug interactions, and eliminate potential dosing errors. Further, GTx-104 has the potential to better manage hypotension in aSAH patients.
|
|
|
• |
GTx-102 is an oral-mucosal betamethasone spray for the treatment of Ataxia Telangiectasia (“A-T”), a complex orphan pediatric genetic neurodegenerative
disorder usually diagnosed in young children, for which no FDA approved treatment currently exists.
|
|
|
• | GTx-101 is a topical bio adhesive film-forming bupivacaine spray for Postherpetic Neuralgia (“PHN”), which can be persistent and often causes debilitating pain following infection by the shingles virus. We believe that GTx-101 could be administered to patients with PHN to treat pain associated with the disease. |
In May 2023, we announced the strategic decision to prioritize development of GTx-104 with a goal to advance the product candidate to commercialization, while conserving resources as much as possible to complete
development efficiently. We estimate that the deferral of GTx-102 and GTx-101 clinical development could be at least three years given the timeline to complete the development and potential commercial launch of GTx-104. Further development of
GTx-102 and GTx-101 will occur at such time as we obtain additional funding or enter into strategic partnerships for license or sale with third parties.
GTx-104
About aneurysmal Subarachnoid Hemorrhage (aSAH)
aSAH is bleeding over the surface of the brain in the subarachnoid space between the brain and the skull, which contains blood vessels that supply the brain. A primary cause of such bleeding is
rupture of an aneurysm. The result is a relatively uncommon type of stroke (aSAH) that accounts for about 5% of all strokes and has an incidence of six per 100,000 person years.
Nimodipine Overview
Nimodipine was granted FDA approval in 1988 and is the only approved drug that has been clinically shown to improve neurological outcomes in aSAH patients. It is only available in the United
States as a generic oral capsule and as a branded oral liquid solution called NYMALIZE™, which is manufactured and sold by Arbor Pharmaceuticals (acquired in September 2021 by Azurity Pharmaceuticals). Nimodipine has poor water solubility and
high permeability characteristics because of its high lipophilicity. Additionally, orally administered nimodipine has dose-limiting side-effects such as hypotension, poor absorption and low bioavailability resulting from high first-pass
metabolism, and a narrow administration window as food effects lower bioavailability significantly. Due to these issues, blood levels of orally administered nimodipine can be highly variable, making it difficult to manage blood pressure in aSAH
patients. Nimodipine capsules are also difficult to administer, particularly to unconscious patients or those with impaired ability to swallow. Concomitant use with CYP3A inhibitors is contraindicated (NIMODIPINE Capsule PI).
NIMOTOP™ is an injectable form of nimodipine that is manufactured by Bayer Healthcare. It is approved in Europe and in other regulated markets (but not in the United States). It has limited
utility for aSAH patients because of its high organic solvent content, namely 23.7% ethanol and 17% polyethylene glycol 400 (NIMOTOP SmPC).
GTx-104 Overview
|
|
• |
GTx-104 is a clinical-stage, novel, injectable of nimodipine for IV infusion in aSAH patients. It uses surfactant micelles as the drug carrier to solubilize nimodipine. This unique nimodipine aqueous
formulation is composed of a nimodipine base, an effective amount of polysorbate 80, a non-ionic hydrophilic surfactant, and a pharmaceutically acceptable carrier for injection. GTx-104 is supplied as an aqueous concentrate that upon
dilution with saline, dextrose, or lactated ringer, is a ready-to-use infusion solution, which is stable and clear.
|
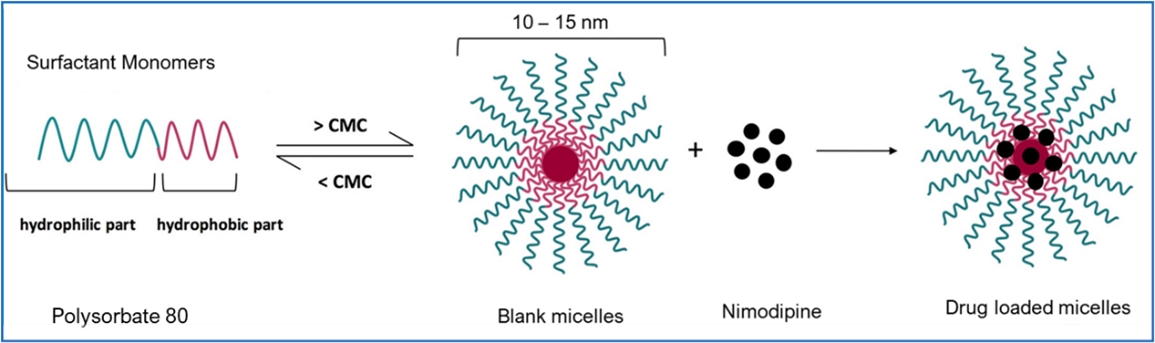
Key potential benefits of GTx-104 include:
|
|
• |
Novel nanoparticle technology facilitates aqueous formulation of insoluble nimodipine for a safe, standard peripheral IV infusion
|
|
|
• |
Better control of blood pressure and improved management of hypotension
|
|
|
• |
100% bioavailability
|
|
|
• |
Eliminates food effects that impact the absorption of the oral form of nimodipine
|
|
|
• |
Lower inter and intra-subject variability as compared to oral nimodipine
|
GTx-104 could provide a more convenient mode of administration as compared to generic nimodipine capsules or NYMALIZE™. GTx-104 is administered as
an IV infusion compared to oral administration via a nasogastric tube in unconscious patients every four hours for both nimodipine capsules and NYMALIZE™. Therefore, GTx-104 could make a major contribution to patient care by potentially
reducing the dosing associated nursing burden. More convenient, continuous, and consistent dosing may also reduce the risk of medication errors. In addition, as depicted in the charts below, the GTx-140-002 PK study conducted by us has shown
that GTx-104 has the potential to provide improved bioavailability and show reduced inter- and intra-subject variability compared to oral nimodipine, which is hypothesized to limit the risk of hypotension and to better achieve desired
therapeutic concentration. Following the capsule administration, the variability was observed higher as compared to IV infusion administration (nimodipine exposure variability at steady state observed 37.5% following oral capsule administration
versus 15.5%, following GTx-104 IV infusion). Because of its IV formulation, we also expect GTx-104 to reduce certain drug-drug interactions and food effects.
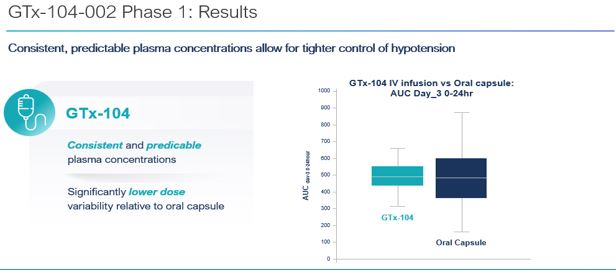
Despite the positive impact it has on recovery, physicians often must discontinue their patients from oral nimodipine, primarily as a result of hypotensive episodes that cannot be controlled by titrating the oral form
of drug. Such discontinuation could potentially be avoided by administering GTx-104, which because of its IV administration, may reduce the complexity associated with the need for careful attention to the timing of nimodipine administration at
least one hour before or two hours after a meal. Also, unconscious patients will likely receive more consistent concentrations of nimodipine when delivered via the IV route as compared to oral gavage or a nasogastric tube. More consistent
dosing is expected to result in a reduction of vasospasm and better, more consistent management of hypotension. As summarized in the table below, we also anticipate reduced use of rescue therapies, such as vasopressors, and expensive hospital
resources, such as the angiography suite, are possible by more effectively managing blood pressure with GTx-104. Reduced incidences of vasospasm could result in shorter length of stay and better outcomes.
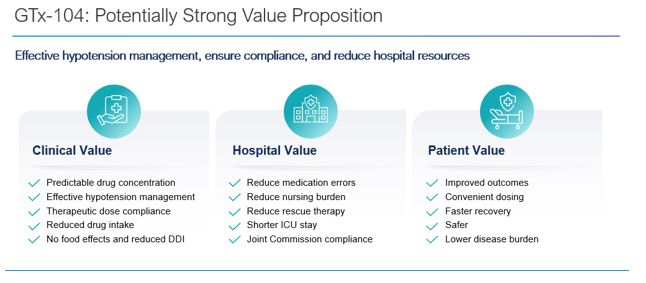
GTx-104 Market Opportunity
Approximately 50,000 patients in the United States are affected by aSAH per year, based on market research. Outside of the United States, annual cases of aSAH are estimated at approximately 60,000
in the European Union, and approximately 150,000 in China.
In contrast to more common types of ischemic stroke in elderly individuals, aSAH often occurs at a relatively young age, with approximately half the affected patients younger than 60 years old. Approximately 10%
to 15% of aSAH patients die before reaching the hospital, and those who survive the initial hours post hemorrhage are admitted or transferred to tertiary care centers with high risk of complications, including rebleeding and delayed cerebral
ischemia (“DCI”). Systemic manifestations affecting cardiovascular, pulmonary, and renal function are common and often complicate management of DCI.
We estimate that total addressable market for aSAH is approximately $300 million in the U.S. There are an estimated 150,000 aSAH patients each
year in China and approximately 55,000 patients in the European Union. The unmet needs in the treatment of aSAH and the potential of GTx-104 to address the limitations of the current standard of care were the subject of a Key Opinion Leader
(“KOL”) event we hosted on October 4, 2023 and will be discussed further at our KOL event planned for November 2024. In an independent market research survey we conducted of hospital administrators, critical and neuro intensive care
physicians at institutions with Comprehensive or Advanced Stroke Center certification who are involved in purchasing decisions for their institutions/units, respondents reported 80% likelihood of adopting an IV formulation of nimodipine
(GTx-104), assuming 100% bioavailability, better safety, no food effects, effective hypotension management, potential hospital value and patient value.
GTx-104 Phase 1 PK Trial
In September 2021, we initiated our pivotal PK bridging trial to evaluate the relative bioavailability of GTx-104 compared to currently marketed oral nimodipine capsules in approximately 50
healthy subjects. The PK trial was the next required step in our proposed 505(b)(2) regulatory pathway for GTx-104.
Final results from this pivotal PK trial were reported in May 2022, and showed that the bioavailability of GTx-104 compared favorably with the oral formulation of nimodipine in all subjects, and
no serious adverse events were observed for GTx-104.
All endpoints indicated that statistically there was no difference in exposures between GTx-104 and oral nimodipine over the defined time periods for both maximum exposure and total exposure.
Plasma concentrations obtained following IV administration showed significantly less variability between subjects as compared to oral administration of capsules, since IV administration is not as sensitive to some of the physiological processes
that affect oral administration, such as taking the drug with and without meals, variable gastrointestinal transit time, variable drug uptake from the gastrointestinal tract into the systemic circulation, and variable hepatic blood flow and
hepatic first pass metabolism. Previous studies have shown these processes significantly affect the oral bioavailability of nimodipine, and therefore cause oral administration to be prone to larger inter- and intra-subject variability.
The bioavailability of oral nimodipine capsules observed was only approximately 8% compared to 100% for GTx-104. Consequently, about one-twelfth
the amount of nimodipine is delivered with GTx-104 to achieve the same blood levels as with the oral capsules. This data is presented in the chart below.
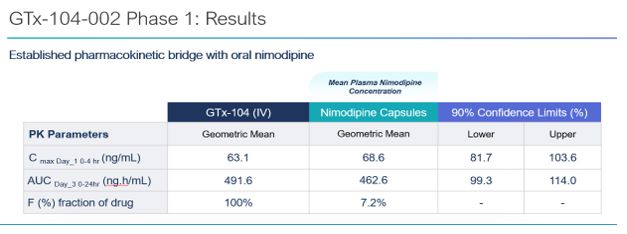
No serious adverse events and no adverse events leading to withdrawal were reported during the trial.
GTx-104 has been administered in over 150 healthy volunteers and was well tolerated with significantly lower inter- and intra-subject pharmacokinetic (“PK”) variability compared to oral
nimodipine.
GTx-104 Pivotal Phase 3 STRIVE-ON Randomized Safety Trial
In April 2023, we received a Type C written meeting response and clarifying feedback from the FDA on our proposed pivotal Phase 3 safety trial for GTx-104. The FDA provided additional comments on
our development plan that, pending submission of the final clinical protocol and FDA approval, would allow us to proceed with a pivotal Phase 3 safety clinical trial in aSAH patients. On July 5, 2023, we announced the alignment with the FDA on
our GTx-104 pivotal Phase 3 safety clinical trial protocol.
The FDA concurred with the suitability of the 505(b)(2) regulatory pathway with the selected Reference Listed Drug NIMOTOP oral capsules (“NDA 018869”), and that our GTx-104-002 PK trial may have
met the criteria for a scientific bridge.
The design of our Phase 3 safety clinical trial, which we have titled STRIVE-ON (Safety, Tolerability, Randomized, IV and Oral Nimodipine; the STRIVE-ON trial–NCT05995405), is a prospective, open-label, randomized (1:1 ratio), parallel group trial of GTx-104 compared with oral nimodipine, in patients hospitalized for aSAH. Key trial design features include:
|
|
• |
Approximately 100 patients will be enrolled at an estimated 25 hospitals in the U.S.
|
|
|
• |
The primary endpoint is safety and will be measured as comparative adverse events, including hypotension, between the two groups.
|
|
|
• |
GTx-104 will be administered as a continuous IV infusion of 0.15 mg/hour, and a 30-minute IV bolus of 4 mg every 4 hours. Oral nimodipine will be administered as 60 mg (two 30 mg capsules) every 4 hours.
|
|
|
• |
Both groups will receive their assigned GTx-104 or oral nimodipine for up to 21 consecutive days and will be evaluated from commencement of patient treatment through a 90-day follow-up period.
|
On October 23, 2023, we enrolled our first patient in the STRIVE-ON trial, and on September 25, 2024, we announced the completion of enrollment.
We anticipate a data readout from the STRIVE-ON trial in first calendar quarter 2025, and plan to submit an NDA to the FDA in the first half of calendar year 2025. If approved, GTx-104 has the potential to address significant challenges with
oral nimodipine administration and may transform the standard of care for patients with aSAH.
GTx-102 Overview
GTx-102 is a novel, concentrated oral-mucosal spray of betamethasone intended to improve neurological symptoms of A-T for which there are currently no FDA-approved therapies. GTx-102 is a
stable, concentrated oral spray formulation comprised of the gluco-corticosteroid betamethasone that, together with other excipients can be sprayed conveniently over the tongue of the A-T patient and is rapidly absorbed.
About Ataxia Telangiectasia
A-T is a rare genetic progressive autosomal recessive neurodegenerative disorder that affects children, with the hallmark symptoms of cerebellar ataxia and other motor dysfunction, and dilated
blood vessels (telangiectasia) that occur in the sclera of the eyes. A-T is caused by mutations in the ataxia telangiectasia gene, which is responsible for modulating cellular response to stress, including breaks in the double strands of DNA.
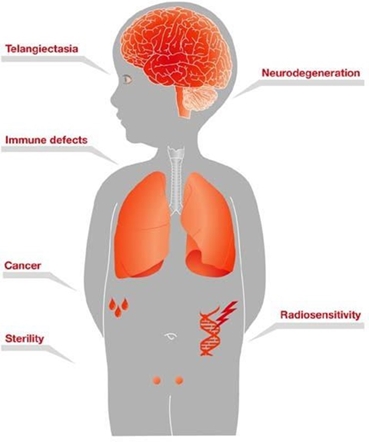
Children with A-T begin to experience balance and coordination problems when they begin to walk (toddler age), and ultimately become wheelchair-bound in their second decade of life. In
pre-adolescence (between ages 5 and 8), patients experience oculomotor apraxia, dysarthria, and dysphagia. They also often develop compromised immune systems and are at increased risk of developing respiratory tract infections and cancer
(typically lymphomas and leukemia).
A-T is diagnosed through a combination of clinical assessment (especially neurologic and oculomotor deficits), laboratory analysis, and genetic testing. There is no known treatment to slow
disease progression, and treatments that are used are strictly aimed at controlling the symptoms (e.g., physical, occupational or speech therapy for neurologic issues), or conditions secondary to the disease (e.g., antibiotics for lung
infections, chemotherapy for cancer, etc.). There are no FDA-approved therapeutic options currently available. Patients typically die by age 25 from complications of lung disease or cancer. According to a third-party report we commissioned, A-T
affects approximately 4,300 patients per year in the United States and has a potential total addressable market of $150 million, based on the number of treatable patients in the United States.
GTx-102 - Research & Development and Clinical Trials to Date
We have licensed the data from the multicenter, double-blinded, randomized, placebo-controlled crossover trial from Azienda Ospedaliera Universitaria Senese, Siena, Italy, where Dr. Zannolli
et. al. studied the effect of oral liquid solution of betamethasone to reduce ataxia symptoms in patients with A-T. This oral liquid solution is not marketed in the United States, and therefore is not available for clinical use. Currently,
betamethasone is only available in the United States as an injectable or as a topical cream. This license gives us the right to reference the trial’s data in our NDA filing. On November 12, 2015, we submitted the data from the Zannolli trial to
the FDA’s Division of Neurology at a pre-Investigational New Drug (“IND”) meeting and received guidance from the agency on the regulatory requirements to seek approval.
In a multicenter, double-blind, randomized, placebo-controlled crossover trial conducted in Italy, Dr. Zannolli et al. studied the effect of an oral liquid solution of betamethasone on the
reduction of ataxia symptoms in 13 children (between ages 2 to 8 years) with A-T. The primary outcome measure was the reduction in ataxia symptoms as assessed by the International Cooperative Ataxia Rating Scale (“ICARS”).
In the trial, oral liquid betamethasone reduced the ICARS total score by a median of 13 points in the intent-to-treat population and 16 points in the per-protocol population (the median percent
decreases of ataxia symptoms of 28% and 31%, respectively). Adverse events in the trial were minimal, with no compulsory withdrawals and only minor side effects that did not require medical intervention. Clinical trial results in A-T patients
administered oral betamethasone indicated that betamethasone significantly reduced ICARS total score relative to placebo (P = 0.01). The median ICARS change score (change in score with betamethasone minus change in score with placebo) was -13
points (95% confidence interval for the difference in medians was -19 to -5.5 points).
Based on the Zannolli data, we believe that our GTx-102 concentrated oral spray has the potential to provide clinical benefits in decreasing A-T symptoms, including assessments of posture and gait
disturbance and kinetic, speech and oculomotor functions. In addition, GTx-102 may ease drug administration for patients experiencing A-T given its application of 1-3x/day of 140µL of concentrated betamethasone liquid sprayed onto the tongue
using a more convenient metered dose delivery system, as these A-T patients typically have difficulty swallowing.
GTx-102 PK Data to Date:
GTx-102 administered as a concentrated oral spray achieves similar blood levels at only 1/70th the volume of an oral solution of betamethasone. This more convenient mode of administration will be
important for A-T patients who have difficulties swallowing large volumes of liquids.
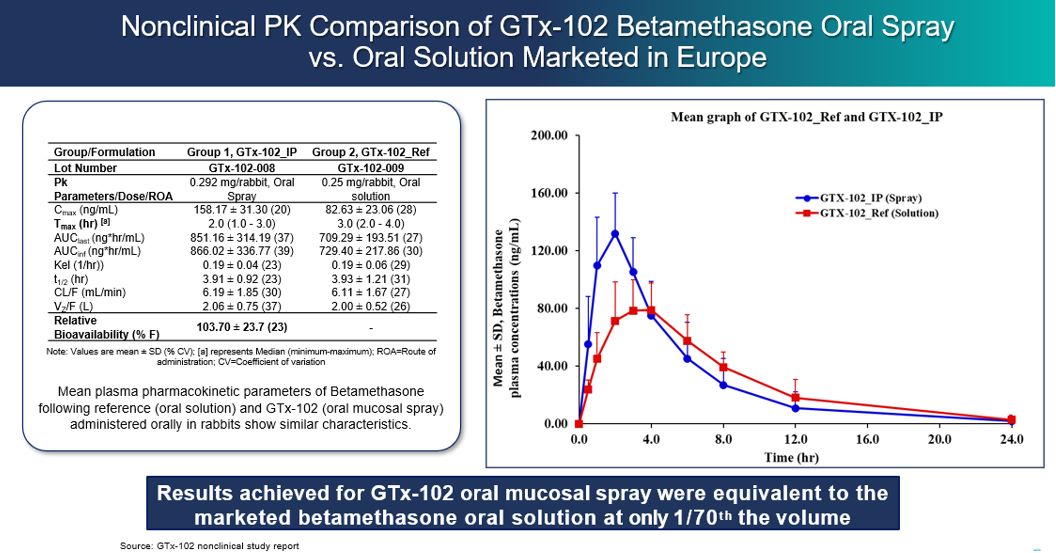
We initiated a PK bridging trial of GTx-102 as compared to the oral liquid solution of betamethasone used in the Zannolli trial and against the injectable form of betamethasone that is approved in the U.S. in the
third calendar quarter of 2022. The primary objectives of the PK bridging trial were to evaluate the bioavailability, pharmacokinetics, and safety of GTx-102. In December 2022, we reported that the topline results of this trial met all primary
outcome measures.
Results showed that GTx-102 betamethasone blood concentrations were highly predictable and consistent based on AUC (the area under the concentration time curve up to 72 hours post-dose, extrapolated
to infinity) and Cmax (the maximum concentration occurring between 0 hour to 72 hours after trial drug administration), indicating good linearity and dose-proportionality. GTx-102 betamethasone blood concentrations were within the same range of
exposure as IM betamethasone, based on AUC. This IM formulation will serve as a bridge for GTx-102 in the context of the proposed 505(b)(2) regulatory pathway. GTx-102 betamethasone blood concentrations were also within the same range of exposure
as Oral Solution (“OS”), based on AUC. This OS formulation was used by Zannolli and may serve as a clinical comparator for further clinical development. Furthermore, statistically there was no significant difference (p>0.05) between GTx-102
administered at a fast rate (each spray immediately following the preceding one) vs. a slow rate (1 spray/minute), as indicated by Cmax and AUC. We believe this result is important because being able to use the fast or the slow rate of
administration may provide greater flexibility for patients and caregivers. The Cmax of GTx-102 was within the same range of exposure as the OS, but the Cmax for the IM formulation was lower than both GTx-102 and the OS, as well as what has been
reported previously for the IM in industry publications. It is important to note that achieving bioequivalence with the IM was not an objective of this trial, nor was it expected. Finally, of the 48 healthy adult subjects, no serious adverse
events were reported, and the most frequent drug-related adverse effect was mild headache (4 cases).
The further clinical development of GTx-102 has been deprioritized in favor of our focus on development of GTx-104. However, we plan to collaborate with clinical experts to design the Phase 3 safety
and efficacy protocol for GTx-102 and gain alignment with the FDA on the development path forward. Further clinical development work will be contingent on additional funding for GTx-102 or the signing of a strategic partnership. It is also
possible that we may license or sell our GTx-102 drug candidate.
GTx-101 Overview
GTx-101 is a non-narcotic, topical bio-adhesive film-forming bupivacaine spray designed to ease the symptoms of patients suffering with postherpetic neuralgia (“PHN”). GTx-101 is administered via a
metered-dose of bupivacaine spray and forms a thin bio-adhesive topical film on the surface of the patient’s skin, which enables a touch-free, non-greasy application. It also comes in convenient, portable 30 ml plastic bottles. Unlike oral
gabapentin and lidocaine patches which are used for the treatment of PHN, we believe that the biphasic delivery mechanism of GTx-101 has the potential for rapid onset of action and continuous pain relief for up to eight hours. No skin sensitivity
was reported in a Phase 1 trial.
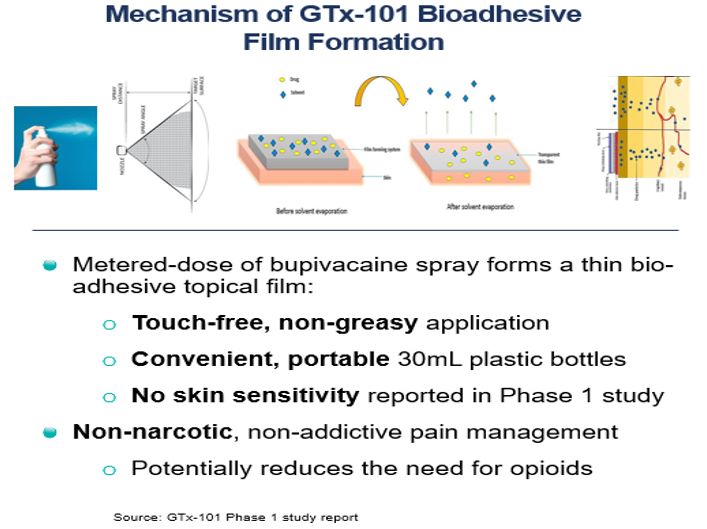
About Postherpetic Neuralgia (PHN)
PHN is neuropathic pain due to damage caused by the varicella zoster virus (“VZV”). Infection with VZV causes two distinct clinical conditions. Primary VZV infection causes varicella (i.e.,
chickenpox), a contagious rash illness that typically occurs among young children. Secondary VZV can reactivate clinically, decades after initial infection, to cause herpes zoster (“HZ”), otherwise known as shingles. Acute HZ arises when dormant
virus particles, persisting within an affected sensory ganglion from the earlier, primary infection with VZV become reactivated when cellular immunity to varicella decreases. Viral particles replicate and may spread to the dorsal root, into the
dorsal horn of the spinal cord, and through peripheral sensory nerve fibers down to the level of the skin. Viral particles also may circulate in the blood. This reactivation is accompanied by inflammation of the skin, immune response, hemorrhage,
and destruction of peripheral and central neurons and their fibers. Following such neural degeneration, distinct types of pathophysiological mechanisms involving both the central and peripheral nervous systems may give rise to the severe nerve
pain associated with PHN.
While the rash associated with HZ typically heals within two to four weeks, the pain may persist for months or even years, and this PHN manifestation is the most common and debilitating
complication of HZ. There is currently no consensus definition for PHN, but it has been suggested by the Centers for Disease Control and Prevention (“CDC”) that PHN is best defined as pain lasting at least three months after resolution of the
rash.
PHN is associated with significant loss of function and reduced quality of life, particularly in the elderly. It has a detrimental effect on all
aspects of a patient’s quality of life. The nature of PHN pain varies from mild to severe, constant, intermittent, or triggered by trivial stimuli. Approximately half of patients with PHN describe their pain as “horrible” or “excruciating,”
ranging in duration from a few minutes to constant on a daily or almost daily basis. The pain can disrupt sleep, mood, work, and activities of daily living, adversely impacting the quality of life and leading to social withdrawal and
depression. PHN is the foremost cause of intractable, debilitating pain in the elderly, and has been cited as the leading cause of suicide in chronic pain patients over the age of 70.
Current treatment of PHN most often consists of oral gabapentin (first line) and prescription lidocaine patches or antidepressants (second line), and refractory cases may be prescribed opioids to
address persistent pain. Gabapentin and opioid abuse have continued to proliferate, and lidocaine patches are suboptimal for many reasons. An independent third-party market research firm we commissioned interviewed more than 250 physicians who
regularly treat PHN patients and found that approximately 40% of patients using lidocaine patches experience insufficient pain relief. Lidocaine patches are difficult to use, fall off, and look unsightly with possible skin sensitivity and
irritation. Additionally, lidocaine patches can only be used for 12 hours and then need to be removed for 12 hours before being reapplied. Prescription lidocaine patches are only approved for PHN, and the market is currently made up of both
branded and generic offerings. It is estimated that PHN affects approximately 120,000 patients per year in the United States. According to a third-party report, the total addressable market for GTx-101 could be as large as $2.5 billion,
consisting of approximately $200 million for PHN pain and $2.3 billion for non-PHN pain indications.
GTx-101 Research & Development History and Clinical Trials Completed to Date
To date, we have conducted four Phase I trials in healthy volunteers to assess the PK, safety, and tolerability of GTx-101 and to determine the plasma levels of bupivacaine HCl administered as a
single dose in various concentrations between 30 mg (three sprays) and 2100 mg (twenty sprays).
These trials confirmed that bupivacaine delivered as a topical spray (GTx-101) is well absorbed through the skin, as demonstrated in the graph below, while very little is absorbed systemically.
In all four trials, the administration of GTx-101 to healthy volunteers was safe and well tolerated. In addition, no evidence of skin irritation was observed at the application site following the
spray administrations. The data below is from two separate trials of GTx-101 and the Lidoderm patch superimposed on each other.
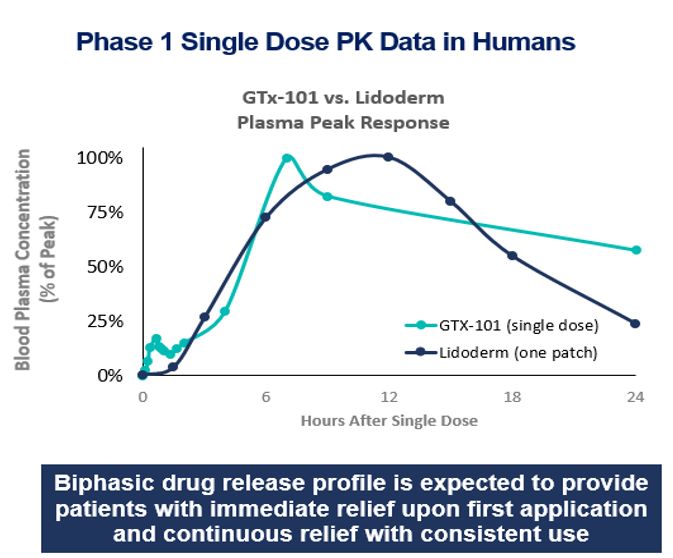
GTx-101 activities:
The data from the single dose Phase 1 clinical trial for GTx-101 was submitted to the FDA’s Division of Anesthesiology and feedback was received at a pre-IND meeting that informed the design of
pre-clinical toxicology studies and a clinical and regulatory pathway to approval under section 505(b)(2). We completed a minipig skin sensitivity study in the second calendar quarter of 2022, and we initiated a single dose PK trial in healthy
human volunteers in July 2022. Topline results from this single dose PK trial were reported in December 2022, and the results met all primary outcome measures.
The median Tmax (the time of maximum concentration between 0 hour and 240 hours after study drug administration) of bupivacaine in plasma following GTx-101 single-dose topical applications ranged
between 18 to 24 hours depending on dose, while the median Tmax following the subcutaneous injection of 10 mg of bupivacaine was only 23 minutes. This result suggests that bupivacaine delivered by GTx-101 remains in the skin for a long period of
time, potentially inducing prolonged analgesic effects in the sprayed area. The exposure to bupivacaine based on Cmax (the maximum concentration occurring at Tmax between 0 hour and 240 hours after study drug administration) and AUC (the area
under the concentration time curve, extrapolated to infinity) following GTx-101 topical application as a single-dose increased with increasing dose.
The systemic exposure to bupivacaine following a 200mg dose of GTx-101 was approximately 29-fold less than a single subcutaneous dose of 10mg of bupivacaine based on Cmax and approximately 6-fold
less than a single subcutaneous dose of 10mg of bupivacaine based on AUC. We predict these lower blood levels will correspond to an increased safety margin for GTx-101 with regards to toxicity risk. Mean half-life (“T half”) following GTx-101
single-dose topical applications ranged between 24 to 37 hours depending on dose, suggesting a slow elimination and potentially long duration of effect, while mean Tmax following the subcutaneous injection of 10 mg of bupivacaine was only 8
hours.
There were only two adverse events judged as related to the study drug by the investigator for each of GTx-101 and the bupivacaine subcutaneous injection. Following GTx-101 topical application:
headache (1 event = 3%) and numbness (1 event = 3%) at the sprayed area following bupivacaine subcutaneous injection: dizziness (1 event = 8%) and nausea (1 event = 8%).
The further development of GTx-101 has been deprioritized in favor of our focus on development of GTx-104. Pending additional funding for GTx-101 or the signing of a strategic partnership, we plan
to follow this successful PK trial with the next step of the clinical development plan including a multiple ascending dose trial. Results from these non-clinical studies and clinical trials are required before the initiation of our Phase 2
program in PHN patients. It is also possible that we may license or sell our GTx-101 drug candidate.
Overall Commercialization Strategy
We have worldwide commercialization rights for all our pipeline drug candidates and plan to maximize the value of each of our drug candidates over time. Currently, we have prioritized the development of GTx-104 over
that of GTx-102 and GTx-101. If we receive regulatory approval for GTx-104 in the US, we may look to out-license its commercialization or consider self-commercialization including outsourcing sales to ensure efficient commercial management and
maximize market penetration and financial returns. We may further seek commercial partnerships to fully exploit the market potential of GTx-104 in territories outside the US. It is possible that we out-license or sell GTx-102 and/or GTx-101 for
the US and/or global markets.
Recent Developments
Completion of Patient Enrollment in Phase 3 STRIVE-ON Safety Trial of GTx-104
As noted above, on September 25, 2024, we announced the completion of enrollment in our Phrase 3 STRIVE-ON trial for GTx-104. We anticipate a
data readout from the STRIVE-ON trial in first calendar quarter 2025 and plan to submit an NDA to the FDA in the first half of calendar year 2025.
Continuance and Domestication
We are a Delaware corporation that, as further described below, previously existed under the laws of the Province of Québec, Canada (“Acasti Québec”), before changing our jurisdiction on October 1, 2024, to the
Province of British Columbia, Canada (“Acasti British Columbia”). On October 7, 2024, we changed our jurisdiction to the State of Delaware (“Acasti Delaware”).
On October 1, 2024, we changed our jurisdiction of incorporation from the Province of Québec in Canada to the Province of British Columbia in
Canada pursuant to a “continuance” effected in accordance with Chapter XII of the Business Corporations Act (Québec) (the “Continuance”). Subsequently on October 7, 2024, we changed our jurisdiction of incorporation from the Province of British
Columbia in Canada to the State of Delaware in the United States of America pursuant to a “continuance” effected in accordance with Section 308 of the Business Corporations Act (British Columbia) and a “domestication” (the “Domestication”)
under Section 388 of the General Corporation Law of the State of Delaware (the “DGCL”). Both the Continuance and the Domestication were approved by our shareholders at our Annual and Special Meeting of Shareholders held on September 30, 2024.
Prior to the Continuance and Domestication, our Class A common shares, without par value per share were listed on The Nasdaq Stock Market LLC
(“Nasdaq”) under symbol “ACST.” Upon the effectiveness of the Continuance, each of our common shares at the time of the Continuance remained issued and outstanding as a common share, without par value per share, of Acasti British Columbia.
Upon effectiveness of the Domestication, each of our outstanding common shares at the time of the Domestication automatically became one outstanding share of common stock, par value $0.0001 per share, of Acasti Delaware. Our common stock
continues to be listed for trading on Nasdaq.
Corporate Name Change
Effective October 28, 2024, we changed our corporate name to Grace Therapeutics, Inc. and our common stock commenced trading under the trading symbol “GRCE” on Nasdaq.
Basis of Presentation of the Financial Statements
Our unaudited condensed consolidated financial statements, which include the accounts of our wholly owned subsidiaries, have been prepared in accordance with U.S. GAAP and the rules and
regulations of the SEC related to quarterly reports filed on Form 10-Q. All intercompany transactions and balances are eliminated on consolidation.
Our assets as of September 30, 2024, include cash, cash equivalents, and short-term investments totaling $15.2 million and intangible
assets and goodwill totaling $49.3 million. Our current liabilities total $2.7 million as of September 30, 2024 and are comprised primarily of amounts due to or accrued for creditors. The Company
believes its cash runway will be sufficient to fund the Company’s operations into the second calendar quarter of 2026.
Results of Operations for the three and the six months ended September 30, 2024 and 2023
|
Three months ended
|
Six months ended
|
|||||||||||||||||||||||
|
September 30,
2024
|
September 30,
2023
|
Increase (Decrease)
|
September 30,
2024
|
September 30,
2023
|
Increase (Decrease)
|
|||||||||||||||||||
|
|
$ |
|
$ |
|
$ |
|
$ |
|
$ |
|
$ | |||||||||||||
|
Operating expenses
|
||||||||||||||||||||||||
|
Research and development expenses, net of government assistance
|
2,976
|
460
|
2,516
|
5,684
|
1,555
|
4,129
|
||||||||||||||||||
|
General and administrative expenses
|
1,855
|
1,632
|
223
|
4,109
|
3,506
|
603
|
||||||||||||||||||
|
Restructuring costs
|
—
|
—
|
—
|
—
|
1,485
|
(1,485
|
)
|
|||||||||||||||||
|
Loss from operating activities
|
(4,831
|
)
|
(2,092
|
)
|
2,739
|
(9,793
|
)
|
(6,546
|
)
|
3,247
|
||||||||||||||
|
|
||||||||||||||||||||||||
|
Foreign exchange gain (loss)
|
13
|
(13
|
)
|
26
|
5
|
(5
|
)
|
10
|
||||||||||||||||
|
Change in fair value of derivative warrant liabilities
|
362
|
(1,826
|
)
|
2,188
|
1,756
|
(1,826
|
)
|
3,582
|
||||||||||||||||
|
Interest and other income, net
|
172
|
212
|
(40
|
)
|
407
|
346
|
61
|
|||||||||||||||||
|
Income tax benefit
|
852
|
446
|
406
|
1,576
|
735
|
841
|
||||||||||||||||||
|
Net loss
|
(3,432
|
)
|
(3,273
|
)
|
159
|
(6,049
|
)
|
(7,296
|
)
|
(1,247
|
)
|
|||||||||||||
Net loss
The net loss of $3.4 million, or $0.30 per share, for the three months ended September 30, 2024, increased by $159 from the net loss of $3.3
million, or $0.43 per share, for the three months ended September 30, 2023. The increase in net loss was primarily due to an increase in research and development expenses of $2.5 million and general and administrative expenses of $223, offset
in part by a $2.2 million difference in the change in the fair value of derivative warrant liabilities and a $0.4 million increase in our income tax benefit.
The net loss of $6.0 million, or $0.53 per share, for the six months ended September 30, 2024, decreased by $1.3 million from the net loss of $7.3
million, or $0.97 per share, for the six months ended September 30, 2023. The decrease in net loss was primarily due to a $3.6 million difference in the change in fair value of derivative warrant liabilities, an $841 increase in our income tax
benefit, and a $1.5 million decrease in restructuring costs, partly offset by a $4.1 million increase in research and development expenses and a $603 increase in general and administrative expenses.
Research and development expenses, net of government assistance
Research and development expenses consist primarily of:
|
|
• |
fees paid to external service providers such as CROs and CMOs related to clinical trials, including contractual obligations for clinical development, clinical sites, manufacturing and
scale-up, and formulation of clinical drug supplies;
|
|
|
• |
fees paid to contract service providers related to drug discovery efforts including chemistry and biology services; and
|
|
|
• |
salaries and related expenses for research and development personnel, including expenses related to stock options.
|
We record research and development expenses as incurred.
Our research and development during the three and the six months ended September 30, 2024 and 2023 was focused primarily on our clinical development program for our GTx-104 drug candidate.
The following table summarizes our research and development expenses:
|
Research and
development
expenses |
||||||||||||||||||||||||
|
Three months ended
|
Six months ended
|
|||||||||||||||||||||||
|
September 30,
2024
|
September 30,
2023
|
Increase (Decrease)
|
September 30,
2024
|
September 30,
2023
|
Increase (Decrease)
|
|||||||||||||||||||
|
|
$ |
|
$ |
|
$ |
|
$ |
|
$ |
|
$ | |||||||||||||
|
Total third-party research and development expenses1
|
2,716
|
152
|
2,564
|
5,150
|
965
|
4,185
|
||||||||||||||||||
|
Government grants & tax credits
|
—
|
4
|
(4
|
)
|
—
|
55
|
(55
|
)
|
||||||||||||||||
|
Salaries and benefits
|
200
|
209
|
(9
|
)
|
408
|
435
|
(27
|
)
|
||||||||||||||||
|
Research and development expense before stock-based compensation and depreciation
|
2,916
|
365
|
2,551
|
5,558
|
1,455
|
4,103
|
||||||||||||||||||
|
Stock-based compensation
|
60
|
83
|
(23
|
)
|
126
|
83
|
43
|
|||||||||||||||||
|
Depreciation and loss on disposal of equipment
|
—
|
12
|
(12
|
)
|
—
|
17
|
(17
|
)
|
||||||||||||||||
|
Total
|
2,976
|
460
|
2,516
|
5,684
|
1,555
|
4,129
|
||||||||||||||||||
1 Total third-party research and development expenses are calculated before salaries and
benefits, depreciation, write-off of equipment and stock-based compensation.
Total research and development expenses for the three and the six months ended September 30, 2024 were $3.0 million and $5.7 million, respectively,
compared to $460 and $1.5 million for the three months and the six months ended September 30, 2023, respectively. This increase of $2.5 million and $4.1 million for the three and the six months period then ended, respectively, was primarily due
to the increase in research activities for the GTx-104 pivotal Phase 3 safety clinical trial.
There were no government grants and tax credits for the three and the six months ended September 30, 2024, compared to $4 and $55 for the three months
and the six months ended September 30, 2023, respectively. The changes within government grants and tax credits were due to adjustments of provisions regarding the estimated realizability of credits receivable after assessments and correspondence
from tax authorities.
Stock-based compensation of $60 for the three months ended September 30, 2024, decreased by $23 compared to $83 for the three months ended September
30, 2023. Stock-based compensation of $126 for the six months ended September 30, 2024, increased by $43 compared to $83 for the six months ended September 30, 2023. The decrease for the three months ended and the increase for the six months
ended, was primarily due to the timing of the issuance of new grants.
General and administrative expenses
General and administrative expenses consist primarily of salaries and related benefits, including stock-based compensation, related to our executive, finance, legal, and support functions,
including professional fees for auditing, tax, consulting, rent and utilities and insurance.
|
General and
administrative
expenses
|
||||||||||||||||||||||||
|
Three months ended
|
Six months ended
|
|||||||||||||||||||||||
|
September 30,
2024
|
September 30,
2023
|
Increase (Decrease)
|
September 30,
2024
|
September 30,
2023
|
Increase (Decrease)
|
|||||||||||||||||||
|
|
$ |
|
$ |
|
$ |
|
$ |
|
$ |
|
$ | |||||||||||||
|
Salaries and benefits
|
490
|
228
|
262
|
885
|
585
|
300
|
||||||||||||||||||
|
Professional fees
|
977
|
864
|
113
|
2,431
|
1,829
|
602
|
||||||||||||||||||
|
Other
|
244
|
340
|
(96
|
)
|
476
|
813
|
(337
|
)
|
||||||||||||||||
|
General and administrative expense before stock-based compensation and depreciation1
|
1,711
|
1,432
|
279
|
3,792
|
3,227
|
565
|
||||||||||||||||||
|
Stock-based compensation
|
142
|
199
|
(57
|
)
|
314
|
275
|
39
|
|||||||||||||||||
|
Depreciation
|
2
|
1
|
1
|
3
|
4
|
(1
|
)
|
|||||||||||||||||
|
Total
|
1,855
|
1,632
|
223
|
4,109
|
3,506
|
603
|
||||||||||||||||||
1 General and administrative sub-total expenses are calculated before stock-based compensation
and depreciation.
General and administrative expenses were $1.9 million and $4.1 million for the three and the six months ended September 30, 2024, respectively, an
increase of $223 and $603, respectively, from $1.6 million and $3.5 million for the three and the six months ended September 30, 2023, respectively. The increase was primarily a result of increased legal, tax, accounting, audit and other
professional fees primarily related to the Continuance and the Domestication, increased salaries and benefits due to merit increases and hiring of new employee, offset by a decrease in other expenses due primarily to adjustments for claims for
Canadian goods and services tax and decrease in miscellaneous expenses as a result of restructuring. Stock-based compensation of $142 and $314 for the three and the six months ended September 30, 2024, respectively, decreased by $57 and increased
by $39, respectively, compared to $199 and $275 for the three and the six months ended September 30, 2023, respectively. The decrease in three months ended and the increase in the six months ended was primarily due to the timing of the issuance
of new grants.
Restructuring Costs
On May 8, 2023, we announced our decision to terminate a substantial amount of our workforce as part of a plan intended to align our organizational and management cost structure to prioritize
resources to GTx-104, thereby reducing losses to improve cash flow and extend available cash resources. We incurred $1.5 million of related costs primarily consisting of employee severance costs. There were no restructuring costs during the three
and the six months ended September 30, 2024.
Change in fair value of derivative warrant liabilities
For the three months ended September 30, 2024, the decrease in the fair value of derivative warrant liabilities of $362 was a mark-to-market
adjustment to the derivative warrant liabilities and primarily due to a decrease in volatility, offset by an increase in our stock price. For the three months ended September 30, 2023, the increase in the fair value of derivative warrant
liabilities of $1.8 million was a mark-to-market adjustment to the derivative warrant liabilities primarily due to an increase in our stock price.
For the six months ended September 30, 2024, the decrease in the fair value of derivative warrant liabilities of $1.8 million was a mark-to-market
adjustment to the derivative warrant liabilities primarily due to a decrease in our stock price. The increase in the fair value of derivative warrant liabilities of $1.8 million for the six months ended September 30, 2023, was a mark-to-market
adjustment to the derivative warrant liabilities primarily attributable to an increase in our stock price.
Interest and other income, net
For the three months ended September 30, 2024, interest and other income was $172, a decrease by $40 compared to $212 for the three months ended
September 30, 2023, primarily due to withdrawals of short-term investments upon their maturity used to fund operations, as well as a decrease in interest rates.
For the six months ended September 30, 2024, interest and other income was $407, an increase of $61 compared to $346 for the six months ended
September 30, 2023, primarily due to the increase in interest earned on average daily balances of cash and cash equivalents.
Income tax benefit
For the three and the six months ended September 30, 2024, income tax benefit was $852 and $1.6 million, respectively, an
increase of $406 and $841, respectively, from $446 and $735 for the three and the six months ended September 30, 2023, respectively, due to net losses recognized by our U.S. subsidiary, which are deemed recoverable to the Company and can be
taken as a benefit over time.
Liquidity and Capital Resources
Cash flows and financial condition for the six months ended September 30, 2024 and 2023
Summary
As of September 30, 2024, cash and cash equivalents were $15.1 million, a net decrease of $7.9 million compared to cash and cash equivalents of $23.0 million at March 31, 2024. We
believe our existing cash and cash equivalents will be sufficient to fund our operations into the second calendar quarter of 2026.
We will require additional capital to fund our daily operating needs beyond that time. We do not expect to generate revenue from product sales unless and until we successfully complete drug
development and obtain regulatory approval, which we expect will take several years and is subject to significant uncertainty. To date, we have financed our operations primarily through public offerings and private placements of our Common
Shares, warrants and convertible debt and the proceeds from research tax credits. Until such time that we can generate significant revenue from drug product sales, if ever, we will require additional financing, which is expected to be sourced
from a combination of public or private equity or debt financing or other non-dilutive sources, which may include fees, milestone payments and royalties from collaborations with third parties. Arrangements with collaborators or others may require
us to relinquish certain rights related to our technologies or drug product candidates. Adequate additional financing may not be available to us on acceptable terms, or at all. Our inability to raise capital as and when needed could have a
negative impact on our financial condition and our ability to pursue our business strategy. We plan to raise additional capital in order to maintain adequate liquidity. Negative results from studies or trials, if any, or depressed prices of our
Common Shares could impact our ability to raise additional financing. Raising additional equity capital is subject to market conditions that are not within our control. If we are unable to raise additional funds, we may not be able to realize our
assets and discharge our liabilities in the normal course of business.
Net cash (used in) operating activities
Net cash used in operating activities for the six months ended September 30, 2024 was $7.8 million, compared to $8.4 million for the six
months ended September 30, 2023, a decrease of $0.6 million. The reduction in net cash used in operating activities during the second quarter of 2025 was primarily due to a $4.2 million increase in research and development activities for our
GTx-104 pivotal Phase 3 STRIVE-ON trial protocol, a $0.6 million increase in general and administrative expenses for legal, tax, accounting and other professional fees related to the Continuance and Domestication, and a $0.3 million increase in
salaries and other benefits, offset by $3.0 million in change in trade and other payables and a $1.5 million decrease in restructuring costs.
Net cash (used in) provided by investing activities
Net cash used in investing activities for the six months ended September 30, 2024, was
from our purchase of short-term investments of $15. Net cash provided by investing activities for the six months ended September 30, 2023, was from the proceeds from the sale of equipment of $110.
Net cash provided by financing activities
There were no financing activities for the six months ended September 30, 2024. Net cash provided by financing activities for the six months ended
September 30, 2023, was primarily attributable to the $7.3 million net proceeds received from the September 2023 private placement offering of our securities.
Private Placement
On September 24, 2023, we entered into a securities purchase agreement (the “Purchase Agreement”) with certain institutional and accredited
investors in connection with a private placement offering of our securities (the “Offering”). Pursuant to the Purchase Agreement, we sold 1,951,371 Common Shares, at a purchase price of $1.848 per Common Share and pre-funded warrants (the
“Pre-funded Warrants”) to purchase up to 2,106,853 Common Shares at a purchase price equal to the purchase price per Common Share less $0.0001. Each Pre-funded Warrant is exercisable for one Common Share at an exercise price of $0.0001 per
Common Share, was immediately exercisable, and will expire once exercised in full. Pursuant to the Purchase Agreement, we also issued to such institutional and accredited investors common warrants (the “Common Warrants”) to purchase Common
Shares, exercisable for an aggregate of 2,536,391 Common Shares. Under the terms of the Purchase Agreement, for each Common Share and each Pre-funded Warrant issued in the Offering, an accompanying five-eighths (0.625) of a Common Warrant was
issued to the purchaser thereof. Each whole Common Warrant is exercisable for one Common Share at an exercise price of $3.003 per Common Share, was immediately exercisable, and will expire on the earlier of (i) the 60th day after the date of
the acceptance by the FDA of an NDA for our product candidate GTx-104 or (ii) five years from the date of issuance. The Offering closed on September 25, 2023. The net proceeds to us from the Offering were approximately $7.3 million, after
deducting fees and expenses.
Contractual Obligations and Commitments
Our contractual obligations and commitments include trade payables, CMO and CRO agreements, and the raw krill oil supply agreement, as described below.
Research and development contracts and contract research organizations agreements
We utilize CMOs for the development and production of clinical materials, and CROs to perform services related to our clinical trials. Pursuant to the agreements with CMOs and CROs, we have either the
right to terminate the agreements without penalties or under certain penalty conditions. At September 30, 2024, the Company has $398 commitment to CMOs and $3.9 million of commitments to CROs for the next twelve months.
Raw krill oil supply contract
On October 25, 2019, we entered into a supply agreement with Aker BioMarine Antarctic AS. (“AKBM”) to purchase raw krill oil product for a committed
volume of commercial starting material for CaPre, one of our former drug candidates, for a total fixed value of $3.1 million based on the value of krill oil at that time. As of March 31, 2022, the remaining balance of commitment amounted to $2.8
million. During the second calendar quarter of 2022, AKBM informed us that AKBM believed it had satisfied the terms of the supply agreement as to their obligation to deliver the remaining balance of raw krill oil product, and that we were
therefore required to accept the remaining product commitment. We disagreed with AKBM’s position and believed that AKBM was not entitled to further payment under the supply agreement. Accordingly, no liability was recorded by us. The dispute
remained unresolved as of both March 31, 2023 and 2022. On October 18, 2023, we entered into an agreement with AKBM to settle any and all potential claims regarding amounts due under the supply agreement (the “Settlement Agreement”). Pursuant to
the terms of the Settlement Agreement, in exchange for a release and waiver of claims arising out of the supply agreement by AKBM and any of AKBM’s affiliates, we agreed to the following: (a) AKBM retained ownership of all raw krill oil product,
including amounts previously delivered to us; (b) AKBM acquired and took ownership of all of our production equipment related to the production of CaPre; (c) AKBM acquired and took ownership of all of our data from research, clinical trials and
pre-clinical studies with respect to CaPre; and (d) AKBM acquired and took ownership over all of our rights, title and interest in and to all intellectual property rights, including all patents and trademarks, related to CaPre owned by us.
Further, AKBM acknowledged that the CaPre assets were transferred on an “as is” basis, and in connection therewith we disclaimed all representations and warranties in connection with the CaPre assets, including any representations with respect to
performance or sufficiency. The value of the raw krill oil previously delivered to us, the production equipment, and the intellectual property rights related to CaPre were fully impaired in prior reporting periods and had a carrying value of nil
as of March 31, 2023. For the three and the six months ended September 30, 2024, there was nil and $193, respectively, in expenses recorded by the Company in relation to shipping cost to transport the Company’s production equipment related to the
production of CaPre.
Contingencies
We evaluate contingencies on an ongoing basis and establish loss provisions for matters in which losses are probable and the amount of the loss can be reasonably estimated.
Use of Estimates and Measurement of Uncertainty
The preparation of these unaudited condensed consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported
amounts of assets, liabilities, income, and expenses. Actual results may differ from these estimates.
Estimates are based on management’s best knowledge of current events and actions that management may undertake in the future. Estimates and underlying assumptions are reviewed on an ongoing
basis. Revisions to accounting estimates are recognized in the period in which the estimates are revised and in any future periods affected.
Estimates and assumptions include the measurement of stock-based compensation, derivative warrant liabilities, accruals for research and development contracts and contract organization
agreements, and valuation of intangibles and goodwill. Estimates and assumptions are also involved in determining which research and development expenses qualify for research and development tax credits and in what amounts. The Company recognizes
the tax credits once it has reasonable assurance that they will be realized.
Critical Accounting Policies
During the six months ended September 30, 2024, there were no material changes to our critical accounting policies from those described in our Annual Report for the year ended March 31, 2024.
Recent Accounting Pronouncements
We have considered recent accounting pronouncements and concluded that they are either not applicable to our business or that the effect is not expected to be material to our consolidated
financial statements as a result of future adoption.
A smaller reporting company is not required to provide the information required by this Item.
Disclosure Controls and Procedures
As of the end of the period covered by this quarterly report, our management, with the participation of our Chief Executive Officer and Principal Financial Officer, has performed an evaluation
of the effectiveness of our disclosure controls and procedures within the meaning of Rules 13a-15(e) and 15d-15(e) of the Exchange Act. Based upon this evaluation, our management has concluded that, as of September 30, 2024, our existing
disclosure controls and procedures were effective. It should be noted that while our Chief Executive Officer and Principal Financial Officer believe that our disclosure controls and procedures provide a reasonable level of assurance that they are
effective, they do not expect the disclosure controls and procedures to be capable of preventing all errors and fraud. A control system, no matter how well conceived or operated, can provide only reasonable, but not absolute, assurance that the
objectives of the control system are met.
Changes in Internal Control over Financial Reporting
No changes were made to our internal controls over financial reporting that occurred during the quarter ended September 30, 2024, that have materially affected, or are reasonably likely to
materially affect, our internal controls over financial reporting.
PART II. OTHER INFORMATION
In the ordinary course of business, we are at times subject to various legal proceedings and disputes. We assess our liabilities and contingencies in connection with outstanding legal
proceedings utilizing the latest information available. Where it is probable that we will incur a loss and the amount of the loss can be reasonably estimated, we record a liability in our unaudited condensed consolidated financial statements.
These legal reserves may be increased or decreased to reflect any relevant developments on a quarterly basis. Where a loss is not probable or the amount of loss is not estimable, we do not accrue legal reserves. While the outcome of legal
proceedings is inherently uncertain, based on information currently available and available insurance coverage, our management believes that it has established appropriate legal reserves. Any incremental liabilities arising from pending legal
proceedings are not expected to have a material adverse effect on our financial position, results of operations, or cash flows. However, it is possible that the ultimate resolution of these matters, if unfavorable, may be material to our
financial position, results of operations, or cash flows. We are not currently a party to any legal proceedings that, in the opinion of management, are likely to have a material adverse effect on our business.
There have been no material changes from the risk factors disclosed in our Annual Report.
None.
None.
Not applicable.
During the three months ended September 30, 2024, no director or officer of the Company adopted terminated
|
Exhibit No.
|
Description
|
|
|
Certificate of Incorporation of Grace Therapeutics, Inc. (incorporated by referenced to Exhibit 3.1 on the Current Report on Form 8-K filed with the Commission on October 7, 2024)
|
|
|
Certificate of Amendment to the Certificate of Incorporation of Grace Therapeutics, Inc. (incorporated by referenced to Exhibit 3.1 on the Current Report on Form 8-K filed with the Commission on October 28,
2024)
|
||
|
|
|
|
|
|
Bylaws of Grace Therapeutics, Inc. (incorporated by reference to Exhibit 3.2 on the Current Report on Form 8-K filed with the Commission on October 28, 2024)
|
|
|
|
|
|
|
10.1†
|
Letter Agreement by and between Prashant Kohli and the Company, dated August 12, 2024 (incorporated by reference to Exhibit 10.1 on the Current Report on Form 8-K filed with the Commission on August 16, 2024)
|
|
|
10.2†
|
Acasti Pharma Inc. 2024 Equity Incentive Plan (incorporated by reference to Exhibit 10.1 on the Current Report on Form 8-K filed with the Commission on September 30, 2024)
|
|
|
10.3†
|
Form of Indemnification Agreement between Acasti Pharma Inc. and its directors and officers (incorporated by reference to Exhibit 10.1 to the Company’s Registration Statement on Form S-4 filed with the
Commission on June 27, 2024)
|
|
|
|
Certification of Chief Executive Officer pursuant to Rule 13a-14(a) or 15d-14(a) of the Securities Exchange Act of 1934
|
|
|
|
|
|
|
|
Certification of Principal Financial Officer pursuant to Rule 13a-14(a) or 15d-14(a) of the Securities Exchange Act of 1934
|
|
|
|
|
|
|
|
Certification of the Chief Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
|
|
|
|
|
|
|
|
Certification of the Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002
|
|
|
|
|
|
|
101.INS
|
|
Inline XBRL Instance Document – the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document
|
|
101.SCH
|
|
Inline XBRL Taxonomy Extension Schema With Embedded Linkbase Documents
|
|
104
|
|
Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101)
|
* Filed or furnished herewith
† Indicates a management contract or compensatory plan.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto
duly authorized.
|
Dated: November 13, 2024
|
||
|
GRACE THERAPEUTICS, INC.
|
||
|
By:
|
/s/ Prashant Kohli
|
|
|
Name: Prashant Kohli
|
||
|
Title: Chief Executive Officer
(Principal Executive Officer)
|
||
|
By:
|
/s/ Robert DelAversano
|
|
|
Name: Robert DelAversano
|
||
|
Title: Principal Financial Officer
(Principal Financial Officer)
|
||
39